Agencia Estatal Boletín Oficial del Estado






 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.Excelentísimos señores:
El Decreto 2519/1974, de 9 de agosto, sobre entrada en vigor, aplicación y desarrollo del Código Alimentario Español, contempla la necesidad de completar la normativa vigente en productos de consumo humano, para lo que es imprescindible la fijación de límites de componentes en la normalización de los diferentes productos, límites que dependen en la mayoría de los casos de las técnicas analíticas a emplear.
Por otra parte, la actuación de los distintos Ministerios en el ámbito de sus respectivas competencias, requiere el análisis de idénticos componentes de los productos de consumo humano, si bien en diferentes fases de elaboración o comercialización.
Por todo lo anterior, parece lógico unificar criterios y aunar esfuerzos mediante el establecimiento de métodos de análisis oficiales únicos para todos los Ministerios.
Para la redacción de los citados métodos de análisis, hecha conjuntamente por cualificados especialistas de los Ministerios interesados, se ha considerado conveniente adaptarse en lo posible a los aprobados por Organismos Internacionales especializados en la materia, con el fin de aprovechar la experiencia habida en su aplicación y de facilitar la confrontación de los resultados en las relaciones comerciales supranacionales.
En consecuencia, a propuesta de los Ministros del Ejército, Hacienda, Gobernación, Industria, Agricultura y Comercio, esta Presidencia del Gobierno dispone:
Se aprueban como oficiales los métodos de análisis de Aceites y Grasas, Cereales y Derivados, Productos Lácteos y Productos Derivados de la Uva que se citan en los anexos 1, 2, 3 y 4.
Cuando no existan métodos oficiales para determinados análisis, y hasta que sean estudiados por el Grupo de Trabajo correspondiente, podrán ser utilizados los adoptados por Organismos nacionales o internacionales de reconocida solvencia.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo digo a VV. EE. a los procedentes efectos.
Dios guarde a VV. EE.
Madrid, 31 de enero de 1977.
OSORIO
Excmos. Sres. Ministros del Ejército, Hacienda, Gobernación, Industria, Agricultura y Comercio.
1. PREPARACIÓN DE LA MUESTRA
1.1 Principio.
Este método establece las condiciones generales de preparación de la muestra; las de carácter particular están indicadas en los métodos correspondientes.
1.2 Procedimiento.
1.2.1 La muestra es fluida y perfectamente limpia.
Para todas las determinaciones, antes de realizar la toma para ensayo, agitar la muestra como medida de precaución.
1.2.2 La muestra es fluida, pero presenta turbidez o materia depositada.
1.2.2.1 Para las determinaciones: Impurezas, agua y materias volátiles, e insaponificable, agitar enérgicamente la muestra para homogeneizarla lo mejor posible antes de la toma para ensayo.
1.2.2.2 Para los demás métodos colocar la muestra en estufa a 50 °C. Cuando aquélla alcance esta temperatura, agitar enérgicamente. Dejar decantar. Filtrar sobre papel en la estufa mantenida a 50 °C. El filtrado debe ser limpio.
1.2.3 La muestra es sólida.
Colocar en estufa mantenida a una temperatura 10 °C superior a la de fusión presumible de la muestra. Operar como en 1.2.1 si la muestra fundida es fluida y perfectamente limpia; y como en 1.2.2 si la muestra fundida presenta turbidez o materia depositada.
1.3 Referencia.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. A. 1.
2. CARACTERES ORGANOLÉPTICOS
2.1 Principio.
Caracteres organolépticos son las cualidades de las sustancias grasas perceptibles directamente por los sentidos. Por lo tanto, su determinación es fundamentalmente subjetiva; no permitiendo establecer, en general, métodos concretos y definidos.
2.2 Aspecto.
Se considerará de aspecto correcto cuando sometida la muestra de aceite, durante 24 horas, a una temperatura de 20 °C ± 2 °C, se observe homogénea, limpia y transparente.
2.3 Olor y sabor.
Serán los normales según el tipo de aceite, y con los aromas propios y característicos, sin que se advierta en ningún caso síntomas organolépticos de rancidez.
2.4 Color.
Variará del amarillo al verde. Para los aceites de oliva y orujo se medirá por el método «Índice de color A. B. T.». En los demás aceites refinados se medirá en el sistema Lovibond, utilizando cubetas de 5,25 pulgadas.
3. ÍNDICE DE COLOR A. B. T.
3.1 Principio.
Este método tiene por objeto establecer una escala de índices para la denominación del color de los aceites de oliva y de semillas, que no contengan, examinados por la visión humana, tonalidades rojizas, es decir, que sólo representen tonalidades variables del amarillo al verde.
El índice de color A. B. T. indica cuantos ml de una disolución 1/15 M de fosfato disódico de Sorensen deberá contener por litro, una mezcla de dicha disolución con otra 1/15 M de fosfato monopotásico, para que agregando un número suficiente de ml de una disolución al 0,04 por 100 de azul de bromotimol, preparada en la forma que se indica más adelante, se origine una coloración idéntica a la del aceite, examinando por transparencia, con la visión humana, una capa de 25 mm de espesor, de la materia grasa y de la disolución patrón.
3.2 Material y aparatos.
3.2.1 Tubos de vidrio neutro de 25 mm de diámetro interior y 140 mm de longitud.
3.3 Reactivos.
3.3.1 Disolución 1/15 M de fosfato monopotásico. Disolver 9,078 g de KH2PO4 en agua destilada y hervida, hasta completar 1 litro.
3.3.2 Disolución 1/15 M de fosfato disódico. Disolver 11,88 gramos de Na2HPO4 2H2O en agua destilada y hervida, hasta completar 1 litro.
3.3.3 Disolución de azul de bromotimol al 0,04 por 100. Triturar en un mortero de ágata 0,1 g de azul de bromotimol, agregar, poco a poco, y removiendo, 3,5 ml de NaOH 0,05N. Cuando se ha disuelto, observándose sólo la turbidez ligera, llevar íntegramente la disolución a un matraz aforado de 250 ml, utilizando para lavar el mortero agua destilada y hervida. Agregar al matraz agua destilada y hervida hasta completar la cuarta parte de su volumen, y calentar en bario de agua a 80°-90 °C, hasta disolución completa. Enfriar hasta la temperatura ambiente, completando con agua destilada y hervida, hasta el enrase.
3.4 Procedimiento.
3.4.1 Preparación de los patrones de color. Poner en cada uno de los nueve tubos de vidrio, los volúmenes de las disoluciones de fosfato monopotásico y disódico que se indican en el cuadro que figura a continuación. Agregar 2 ml de la disolución de azul de bromotimol y agitar los tubos.
| Indice A. B. T. | Disolución 1/15 M en ml KH, PO. |
Disolución 1/15 M en ml. Na2HPO, 2H2O |
|---|---|---|
| 0 | 50,00 | 0,00 |
| 25 | 48,75 | 1,25 |
| 50 | 47,50 | 2,50 |
| 75 | 46,25 | 3,75 |
| 100 | 45,00 | 5,00 |
| 125 | 43,75 | 6,25 |
| 150 | 42,50 | 7,50 |
| 175 | 41,25 | 8,75 |
| 200 | 40,00 | 10,00 |
En esta escala el índice O corresponde al patrón con coloración amarilla y el 200 al de la verde, presentando los intermedios, tonos verdosos ascendentes del 0 al 200.
Si es necesario preparar otras series con las mismas mezclas de fosfatos, pero poniendo volúmenes mayores o menores de azul de bromotimo, para obtener intensidades más fuertes o más débiles del tono normal que se fija en este método. Designar estos nuevos índices colocando entre paréntesis, a continuación de los que se establecen en este método, el número de ml de azul de bromotimol utilizados.
Estos patrones se conservan mucho tiempo en la oscuridad, bastando en general una comprobación cada 6 meses, por comparación con disoluciones recién preparadas.
3.4.2 Determinación del índice de color, El aceite cuyo color se quiere describir debe tener una temperatura aproximada de 20 °C y estar completamente transparente, filtrándose si presenta turbidez.
Llenar de aceite hasta las tres cuartas partes uno de los tubos; observar por transparencia, mirando en dirección normal al eje del tubo, con cuál de los colores de la escala de patrones se identifica, colocando detrás de los tubos una hoja de papel blanco.
3.5 Expresión de resultados.
3.5.1 El índice de color se expresará por un número, correspondiente a los ml del patrón con el que se ha identificado la muestra.
3.6 Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.021.
4. DENSIDAD
4.1 Principio.
Se determina la masa de la unidad de volumen, expresada en gramos por centímetro cúbico, a una temperatura dada. La densidad se representa por d.
La temperatura se ha de controlar exactamente ya que la densidad de las materias grasas varía aproximadamente 0,00068 por grado.
La temperatura de la determinación no diferirá de la de referencia en más de 5 °C.
4.2 Material y aparatos.
4.2.1 Picnómetro normal, o con termómetro acoplado de 50 ml aproximadamente.
4.3 Procedimiento.
4.3.1 Aceites y grasas líquidas. Para la determinación de la densidad el picnómetro ha de estar a la temperatura constante del medio ambiente. Llenar el picnómetro hasta el borde superior del tubo capilar, introducir el termómetro, pesar y anotar la temperatura de la determinación.
4.3.2 Grasas sólidas. Llenar el picnómetro hasta las tres cuartas partes, aproximadamente, de su altura, con la grasa. Dejar 1 hora en estufa, a la temperatura de fusión de la grasa, enfriar, pesar. Añadir agua, a la temperatura de referencia, hasta el borde superior del picnómetro, dejar 1 hora en un baño a la temperatura de referencia, secar el picnómetro y pesar.
4.4 Cálculo.
Calcular la densidad expresada en g/cm3 y referida a una temperatura que generalmente será de 20 °C para los aceites y de 40°, 60 °C, etc., para las grasas sólidas.
4.4.1 Aceites y grasas líquidas.

4.4.2 Grasas sólidas.

P = peso en g del picnómetro vacío.
P' = peso en g del picnómetro lleno con agua a la temperatura de referencia.
P" = peso en g del picnómetro lleno con aceite a la temperatura de referencia.
p" = peso en g del picnómetro lleno con grasa y agua a la temperatura de referencia.
D = densidad del agua a la temperatura de la determinación (tabla 4.I).
4.4.3 Correcciones.
El valor de la densidad calculado anteriormente puede corregirse del efecto del empuje del aire por la fórmula
Densidad corregida = d + 0,0012 (1 ‒ d)
d = densidad sin corregir.
4.5 Observaciones.
La temperatura de la determinación y la temperatura de referencia se relacionan en la siguiente forma:
d' = d + (t ‒ t') 0,00068 si t > t
d' = d ‒ (t'' ‒ t) 0,00068 si t < t
d = densidad a la temperatura de la determinación t.
d' = densidad a la temperatura de referencia t.
4.6 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1954.
2. Consejo Oleícola Internacional, 1967.
TABLA 4.I
Densidad del agua en función de la temperatura, tomando como unidad la masa de 1 ml de agua a 4 °C
| 0° 0,999868 | 11° 0,999637 | 21° 0,998019 |
| 1° 0,999927 | 12° 0,999525 | 22° 0,997697 |
| 2° 0,999968 | 13° 0,999404 | 23° 0,997565 |
| 3° 0,999992 | 14° 0,999271 | 24° 0,997323 |
| 4° 1,000000 | 15° 0,999126 | 25° 0,997071 |
| 5° 0,999992 | 16° 0,998970 | 26° 0,996810 |
| 6° 0,999968 | 17° 0,998801 | 27° 0,996539 |
| 7° 0,999929 | 18° 0,998622 | 28° 0,996259 |
| 8° 0,999876 | 19° 0,998432 | 29° 0,995971 |
| 9° 0,999808 | 20° 0,998230 | 30° 0,995673 |
| 10° 0,999728 |
Los valores dados son numéricamente iguales a la densidad absoluta en g/ml.
5. PRUEBA DEL FRÍO
5.1 Principio.
Este método mide la resistencia de la muestra a la cristalización y se usa corrientemente como un índice de los procesos de desmargarización.
Es aplicable a todos los aceites vegetales y animales refinados y secos.
5.2 Material y aparatos.
5.2.1 Frascos de vidrio de unos 115 ml, limpios y secos.
5.2.2 Baño de agua y hielo troceado. Llenar un recipiente de 2 ó 3 litros de capacidad con hielo finamente machacado, y añadir agua fría en cantidad suficiente para que quede cubierto el cierre del frasco que contenga la muestra.
5.3 Procedimiento.
Filtrar una cantidad suficiente de muestra (200 a 300 ml) a través de papel de filtro. Calentar el filtrado, agitándolo continuamente hasta que adquiera exactamente una temperatura de 130 °C.
Llenar completamente un frasco con el aceite muestra filtrado y tapar suavemente con un tapón de corcho. Llevar a 25 °C en un baño de agua y recubrir el tapón con parafina.
Sumergir el frasco en el baño agua-hielo, de forma que quede cubierto el cierre de aquél. Reponer hielo para mantener los 0 °C y el nivel primitivo.
Al cabo de cinco horas y media retirar el frasco del baño y examinarlo detenidamente para ver si se han formado cristales o enturbamiento. No confundir las burbujas de aire finamente dispersadas con los cristales de grasa. La muestra habrá resistido la prueba si se conserva clara, limpia y brillante.
5.4 Expresión de resultados.
Negativa o positiva.
5.5 Observaciones.
El fin del calentamiento inicial es eliminar las trazas de humedad y destruir los núcleos cristalinos que pueden existir. Ambos interferirían la prueba ocasionando enturbiamiento por cristalización prematura.
5.6 Referencias.
1. American Oil Chemists' Society. Official and Tentativa Methods. Cc 11-42.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.042.
6. ÍNDICE DE REFRACCIÓN
6.1 Principio.
El índice de refracción de una sustancia dada es la razón de la velocidad de un rayo de luz en el vacío a la velocidad de luz a través de la sustancia. Por conveniencia práctica se refiere a la relación aire-sustancia.
Es igualmente la relación del seno del ángulo de incidencia al seno del ángulo de refracción.
El índice de refracción de una sustancia dada varía con la longitud de onda del rayo de luz refractado y con la temperatura. Salvo indicación contraria el índice de refracción viene referido a la longitud de onda correspondiente a la línea D 589,3 nm de la luz del sodio. El índice de refracción se indica con la notación nt para C y longitud de onda de la línea D del sodio. Para otra radiación de distinta longitud de onda a t °C, la notación será ntλ.
Con los refractómetros usuales la observación se hace en luz difusa, provistos de un dispositivo de acromatismo de la radiación D de la luz del sodio.
6.2 Material y aparatos.
6.2.1 Refractómetro de precisión, que permita apreciar como mínimo las diezmilésimas, con prismas calentados por circulación de líquido termostatado, ± 0,1 °C. Puede usarse una luz blanca si el refractómetro utilizado posee un dispositivo de compensación cromática.
6.3 Procedimiento.
El aceite debe estar limpio y exento de agua. Filtrar sobre papel de filtro seco, con la ayuda, si es necesario, de sulfato sódico anhidro. Llenar con la materia grasa el espacio comprendido entre los dos prismas. Hacer la lectura después de 5 minutos, al menos, de contacto. La temperatura de lectura no debe sobrepasar en ± 2° la temperatura de referencia.
6.4 Cálculo.
Calcular el índice de refracción referido a la temperatura de 20° para las grasas líquidas a esa temperatura, y referido a 40°, 60°, 80° o temperaturas superiores para las materias grasas sólidas.
nt = nt' + (t' ‒ t) F si t' > t
nt = nt' + (t ‒ t') F si t' < t
nt = índice de refracción a la temperatura de referencia tº.
nt' = índice de refracción a la temperatura de lectura t'°.
F = factor de corrección por temperatura.
F = 0,00035 para t = 20°
F = 0,00036 para t = 40° o superior.
6.5 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II-B.2.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.015.
7. PUNTO DE FUSIÓN
7.1 Principio.
Las grasas y aceites naturales, como mezclas de glicéridos y otras sustancias no tienen punto de fusión neto y definido. No presentan punto crítico de sólido a líquido; este paso lo realizan gradualmente a través de estados pastosos hasta el completamente líquido.
Por tal razón el punto de fusión de una grasa viene definido en este método por dos temperaturas: Una, la inicial de ablandamiento deslizante, y otra, final de líquido perfectamente limpio.
7.2 Material y aparatos.
7.2.1 Tubo de vidrio en U de 1,4-1,5 mm de diámetro, con espesor de pared de 0,15-0,20 mm. Una de las ramas de 80 milímetros de largo, y la otra de 60 mm (ésta ligeramente abocada en el extremo). Distancia entre las dos ramas, 5 mm aproximadamente.
7.2.2 Baño de agua exenta de aire con dispositivo de calefacción lenta.
7.2.3 Termómetro hasta 70 °C, graduado en décimas de grado.
7.3 Procedimiento.
La determinación exacta de las dos temperaturas, inicial y final, depende de las condiciones en que se realice la solidificación de la muestra.
Introducir la rama más larga del tubo en la grasa fundida a una temperatura de 10 °C superior al punto de fusión presumible. Deslizar la columna de grasa tomada hasta 1 cm del codo del tubo; la columna de grasa será aproximadamente de 1 cm de largo.
La grasa puede también introducirse al estado sólido en el tubo, con ayuda de un hilo de platino.
Dejar enfriar el tubo con la grasa durante 24 horas a la temperatura ambiente (por lo menos 10° más baja que el punto inicial de ablandamiento).
Acoplar el tubo a lo largo del termómetro mediante un anillo de goma, de forma que su curvatura coincida con el bulbo del termómetro. Introducir termómetro y tubo en el baño de agua; cuidar que el nivel de ésta sea inferior al de la rama más corta.
Calentar lentamente, a razón de 0,1-0,2 °C por minuto, observando con una lupa la columna de grasa, bien iluminada sobre fondo oscuro. La temperatura inicial es aquella en que la columna comienza a descender (se descuelga). La temperatura final corresponde a la desaparición de todo enturbiamiento y aspecto limpio.
En caso de duda del punto final se compara el tubo de ensayo con otro con grasa netamente fundida.
7.4 Expresión de los resultados.
Para utilización técnica de resultados puede ser suficiente la indicación de la temperatura del punto inicial.
Pero si se utiliza sólo la expresión: «Punto de fusión» se indicarán las dos temperaturas, inicial y final.
Si la grasa se ha introducido en el tubo al estado sólido se hará constar: «Punto de fusión» (sin fundir previamente la grasa).
7.5 Referencia.
1. American Oil Chemists' Society. Official and Tentativa Methods. Cc 1-25.
8. HUMEDAD
(Método del Xilol)
8.1 Principio.
Este método determina la cantidad total de agua no combinada que se encuentra en la materia grasa.
8.2 Material y aparatos.
8.2.1 Matraz de vidrio de cuello corto, de 300 a 500 ml de capacidad, sobre el cual se adapta el aparato especial representado en la figura 8.1. Consta de un tubo cilíndrico graduado en ml, provisto de llave de descarga en el extremo inferior; en el superior se adapta un refrigerante de reflujo. Entre éste y la terminación de la graduación, lleva el tubo cilíndrico otro tubo comunicante y paralelo a él, a cuyo extremo se adapta el matraz.
8.3 Reactivos.
8.3.1 Xileno.
8.4 Procedimiento.
Eliminar todo vestigio de grasa del tubo graduado y del tubo interior del refrigerante, lavando sucesivamente con mezcla crómica, agua destilada y acetona. Secar.
Pesar de 20 a 50 g de materia grasa, en el matraz seco, con una aproximación de 0,1 g. Agregar de 100 a 300 ml de xileno y algunos trozos de piedra pómez. Calentar progresivamente hasta la ebullición y mantenerla hasta que el xileno destilado resulte limpio y no separe más agua. Dejar en reposo hasta perfecta separación de las capas de xileno y agua. Leer el volumen de agua.
8.5 Cálculo.
Calcular el contenido de agua expresado en porcentaje.

P = peso en g de la muestra.
V = volumen en ml de agua.
8.6 Observaciones.
Si las gotas de agua quedan adheridas a la pared del tubo, desprenderlas calentando con precaución con una llama pequeña.
8.7 Referencias.
1. E. W. Dean. D. A. Stark. Ind. Eng. Chem. 1920. 12. 486.
2. American Oil Chemists' Society. Official and Tentativa Methods. Ca. 2a-45.
3. Instituto de Racionalización del Trabajo. Una Norma Española 55.001.
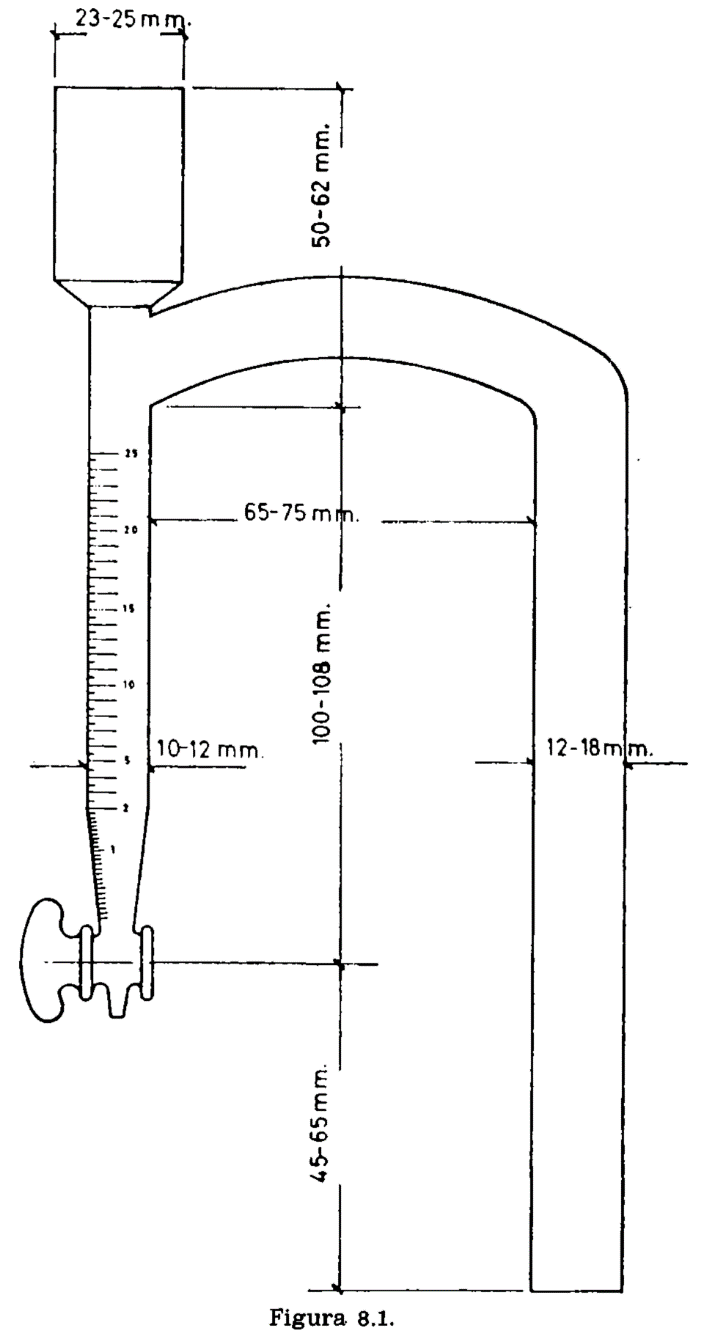
9(a). HUMEDAD Y MATERIAS VOLÁTILES
(Método de la estufa de aire)
9(a).1 Principio.
Se establecen las condiciones adecuadas para la determinación, en las materias grasas, del agua y de las materias volátiles, operando en las condiciones del ensayo.
Es aplicable a las grasas animales y vegetales, con la excepción de los aceites secantes o semisecantes y los aceites del grupo del coco.
9(a).2 Material y aparatos.
9(a).2.1 Estufa de desecación, con regulación de temperatura, pudiéndose calentar hasta 150 °C como mínimo; la regulación se efectuará entre unos límites de oscilación de ± 2 °C, siendo, además, la temperatura uniforme en todo el espacio interior tolerándose diferencias que no excedan de 1 °C entre posiciones extremas.
9(a).2.2 Cápsulas de fondo plano, con dimensiones aproximadas de 80 mm de 0 y 20 mm de altura, preferiblemente de acero inoxidable o aluminio o, en su defecto, de porcelana.
9(a).2.3 Desecador, conteniendo como agente desecante sulfato cálcico o gel de sílice de 6-20 mallas, o, en su defecto, ácido sulfúrico monohidrato (d = 1,84), aunque para asegurar una desecación efectiva deberá ser renovado con frecuencia.
9(a).3 Procedimiento.
9(a).3.1 Preparación de la muestra.
La muestra debe ser previamente homogeneizada antes de pesar la cantidad con que se vaya a operar. Esto se logra, con las grasas fluidas, agitando fuertemente el frasco que contiene la muestra y vertiendo rápidamente la cantidad aproximada que se vaya a pesar en la cápsula en la que se efectúe la desecación. Si se tratase de grasas sólidas o semisólidas a la temperatura ambiente, calentar suavemente en baño de agua hasta conseguir el grado de fluidez conveniente, cuidando de no llegar a fundir completamente y homogeneizar con un mezclador adecuado o simplemente con una espátula si no se dispusiese de este instrumento.
9(a).3.2 Técnica operatoria.
En una cápsula, desecada previamente en estufa a 105 °C y enfriada en un desecador, pasar, con exactitud de 1 miligramo, una cantidad aproximada de 5 a 10 g de muestra., según el contenido de humedad, preparada como se indica en 9(a).3.1.
Colocar en la estufa, previamente regulada a 105 °C, manteniéndola allí durante 30 minutos. Sacar y pasar a un desecador donde se deja enfriar; pesando a continuación. Repetir este tratamiento, en operaciones sucesivas, hasta que la diferencia entre dos pesadas consecutivas no exceda del 0,05 por 100.
9(a).4 Cálculos.

Siendo:
Pa = peso, en gramos, de la cápsula con la muestra de grasa.
Pf = peso, en gramos, de la cápsula con la grasa al dar por terminada la desecación.
PM = peso, en gramos, de la muestra.
9(a).5 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.020.
2. International Union of Pure and Applied Chemistry Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. C.1.1.
9(b). HUMEDAD Y MATERIAS VOLÁTILES
(Método de la estufa de vacío)
9(b).1 Principio.
Se establecen las condiciones adecuadas para la determinación del agua y las materias volátiles, en las condiciones del ensayo, en materias grasas comerciales.
Es aplicable a todos los aceites y grasas del comercio, incluyendo los aceites secantes y semisecantes. No es aplicable a los aceites del grupo del coco, conteniendo un 1 por 100 o más de ácidos grasos libres, ni a grasas que hayan sido adicionadas de monoglicéridos.
9(b).2 Material y aparatos.
9(b).2.1 Estufa de vacío. Estufa en cuyo interior pueda hacerse el vacío, bien sea con trompa de agua o con bomba de características adecuadas, pudiéndose mantener el vacío con la estufa desocupada, al límite máximo alcanzable con la trompa de agua o 1 mm de Hg como mínimo, si se opera con bomba de aceite. En las operaciones de desecación es preferible operar con trompa de agua.
Irá provista de dispositivos de calefacción y regulación de temperatura, pudiéndose calentar hasta 110 °C como mínimo, regulándose entre unos límites de oscilación de ± 2 °C; la temperatura será, además, uniforme en todo el espacio interior, tolerándose diferencias que no superen 1 °C entre posiciones extremas.
La estufa irá provista de termómetro, convenientemente calibrado, dispuesto de la forma conveniente para que el operador pueda comprobar, durante su funcionamiento, la temperatura que existe en el interior.
9(b).2.2 Cápsulas de fondo plano, con dimensiones aproximadas de 80 mm de diámetro, de 20 mm de altura, de acero inoxidable, aluminio o porcelana.
9(b).2.3 Desecador, conteniendo como agente desecante sulfato cálcico o gel de sílice de 6-20 mallas. También puede ser utilizado el ácido sulfúrico monohidrato (d = 1,84), aunque para asegurar una desecación efectiva deberá ser renovado con frecuencia.
9(b).3. Procedimiento.
9(b).3.1 Preparación de la muestra.
La muestra debe ser previamente homogeneizada antes de pesar la cantidad con que se vaya a operar. Esto se logra, con las grasas fluidas, agitando fuertemente el frasco que las contiene y vertiendo rápidamente la cantidad aproximada que se vaya a pesar en la cápsula en la que se efectúe la desecación. Si se tratase de grasas sólidas o semisólidas a la temperatura ambiente, calentar suavemente en baño de agua hasta conseguir el grado de fluidez conveniente, cuidando de no llegar a fundir completamente y homogeneizar con un mezclador adecuado o simplemente con una espátula si no se dispusiese de este instrumento.
9(b).3.2 Técnica operatoria.
En una cápsula desecada previamente en estufa a 105 °C y enfriada en un desecador, pesar, con exactitud de medio miligramo, una cantidad aproximada de 5 a 10 g de muestra, según el contenido de humedad, preparada como se ha indicado en 9(b).3.1.
Colocar en la estufa y hacer el vacío debiéndose alcanzar una presión interna que no sea superior a 100 mm Hg. Poner en marcha la calefacción, regulándose a la temperatura en relación a la presión interna de la estufa. Las condiciones normales de trabajo deben ser 50 mm Hg y 60 °C; para una presión de 100 mm Hg, la temperatura será de 75 °C; para valores intermedios, se calculará mediante interpolación entre las cifras indicadas.
Mantener la cápsula en la estufa durante una hora. Sacar y pasar a un desecador donde se deja enfriar, pesando a continuación. Repetir este tratamiento en operaciones sucesivas, hasta que la diferencia entre dos pesadas consecutivas no exceda del 0,05 por 100.
9(b).4 Cálculos.

Po = peso, en gramos, de la cápsula vacía y seca.
PA = peso, en gramos, de la cápsula con la muestra de grasa.
PF = peso final, en gramos, de la cápsula con la grasa, al dar por terminada la desecación.
9(b).5. Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.082.
10. ACIDEZ ÍNDICE DE ACIDEZ
10.1 Principio.
La acidez que figura normalmente en los boletines de análisis es una expresión convencional del contenido en tanto por ciento de los ácidos grasos libres. También se denomina grado de acidez.
Índice de acidez, expresa el peso, en mg, de hidróxido potásico necesario para neutralizar un gramo de materia grasa.
10.2 Reactivos.
10.2.1 Solución etanólica de hidróxido potásico 0,5 N ó 0,1 N. 10.2.2. Solución al 1 por 100 de fenolftaleína en metanol de 95 por 100 v/v.
10.2.3 Mezcla etanol-éter etílico, 1:1, neutralizada exacta-menta con KOH 0,1 N etanólica, con fenolftaleína como indicador.
10.3 Procedimiento.
Pesar con una aproximación de 0,01 g, 5 a 10 g de grasa, en un erlenmeyer de 250 ml. Disolverla en 50 ml de la mezcla etanol-éter etílico. Valorar, agitando continuamente, con KOH 0,5 N (o con 0,1 N para acideces inferiores a 2), hasta viraje del indicador.
10.4 Cálculo.
Calcular la acidez como grado de acidez expresado en porcentaje de ácido oleico o como índice de acidez expresado en mili-gramos de KOH.


V = volumen en ml de solución etanólica de KOH utilizada.
N = normalidad exacta de la solución de KOH utilizada.
M = peso molecular de ácido en que se expresa la acidez.
P = peso en gramos de la muestra utilizada.
Normalmente se expresa referida a tanto por ciento de ácido oleico Sólo en casos particulares, según la naturaleza de la sustancia grasa, se expresará referida a ácido palmítico, láurico u otros.
10.5 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964.II.D.I.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.011.
3. Consejo Oleícola Internacional, 1967.
11. ÍNDICE DE SAPONIFICACIÓN
11.1 Principio.
El índice de saponificación expresa el peso en mg de hidróxido potásico necesario para saponificar 1 g de grasa.
Este método es aplicable a aceites y grasas con un contenido de ceras inferior al 5 por 100.
11.2 Material y aparatos.
11.2.1 Matraz de vidrio, inatacable por los ácidos, de 200 ml aproximadamente, adaptable a un refrigerante de reflujo.
11.3 Reactivos.
11.3.1 Solución etanólica de hidróxido potásico 0,5 N.
11.3.2 Solución acuosa de ácido clorhídrico 0,5 N.
11.3.3 Solución de fenolftaleína al 1 por 100 en etanol de 95°.
11.4 Procedimiento.
Pesar con una precisión de 1 mg, en el matraz de vidrio, 2 g aproximadamente de grasa. Agregar 25 ml exactamente medidos de solución etanólica de KOH 0,5 N. Adaptar el refrigerante de reflujo, llevar a ebullición, y mantener durante 60 minutos, agitando por rotación de cuando en cuando. Retirar de la fuente de calor. Agregar 4 ó 5 gotas de fenolftaleína, y valorar la solución jabonosa, todavía caliente, con la solución de ácido clorhídrico 0,5 N.
Realizar en las mismas condiciones un ensayo en blanco
11.5 Cálculo.
Calcular el índice de saponificación expresado en mg de KOH por g de grasa.

V = volumen en ml de solución de ClH 0,5 N utilizados en la prueba en blanco.
V' = volumen en ml de solución de ClH 0,5 N utilizados en el ensayo.
N = normalidad exacta de la solución de ácido clorhídrico utilizado.
P = peso en g de la muestra de grasa.
11.6 Observaciones.
Para ciertas materias grasas difíciles de saponificar es necesario calentar durante más de 60 minutos.
11.7 Referencias.
1. Kóttstorfer J. Zeitschrift F. Anal. Chemie. 1879. 18, 199.
2. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps 1964. II. D. 2.
3. Instituto de Racionalización del Trabajo. Una norma española 55.012.
4. Consejo Oleícola Internacional, 1967.
12. ÍNDICE DE HIDRÓXILO
12.1 Principio.
El índice de hidróxilo es el peso en mg de hidróxido potásico necesario para neutralizar el ácido acético que se combina por acetilación con 1 g de grasa.
12.2 Material y aparatos.
12.2.1 Dos matraces de vidrio de 150 ml con cuello de 55 milímetros de largo y diámetro interior de 20 mm.
12.2.2 Pequeños embudos de vidrio de 45 mm de diámetro y 50 mm de vástago.
12.2.3 Placas de cartón, circulares, del diámetro de los matraces con un orificio circular en el centro de diámetro adaptable al cuello de los matraces.
12.2.4 Bario de glicerina, que permita colocar los matraces en posición inclinada, introducidos 1 cm en el baño, protegidos los cuellos por el cartón que apoya en el borde exterior del baño.
12.3 Reactivos.
12.3.1 Anhídrido acético puro.
12.3.2 Piridina pura y seca.
12.3.3 Etanol 95 por 100, neutro a la fenolftaleína.
12.3.4 Solución alcohólica de hidróxido potásico 0,5 N.
12.3.5 Disolución alcohólica de fenolftaleína al 1 por 100.
12.3.6 Reactivo piridina-anhídrido acético. Introducir 25- mililitros de piridina en un matraz aforado de 100 ml, disolver-los en 25 g de anhídrido acético; homogeneizar y enrasar a 100 ml con piridina. Conservar este reactivo al abrigo de la humedad, del anhídrido carbónico o de los vapores ácidos, en frascos de color topacio, bien tapados.
12.4 Procedimiento.
Pesar en el matraz perfectamente seco, con una aproximación de 1 mg, la cantidad de grasa necesaria, según el cuadro siguiente:
| Índice previsto |
Peso de muestra g |
Vol. de reactivo ml |
|---|---|---|
| 10 - 100 | 2,0 | 5 |
| 100 - 150 | 1,5 | 5 |
| 150 - 200 | 1,0 | 5 |
| 200 - 250 | 0,75 | 5 |
| 250 - 300 | 0,60 | 5 |
| 1,2 | 10 | |
| 300 - 350 | 1,0 | 10 |
| 350 - 700 | 0,75 | 15 |
| 700 - 950 | 0,50 | 15 |
| 950 - 1.500 | 0,30 | 15 |
| 1.500 - 2.000 | 0,20 | 15 |
Agregar con una bureta el volumen de reactivo.
Regular el bario de glicerina a 95° - 100 °C. Colocar el disco de cartón alrededor del cuello del matraz. Situar perfectamente un embudo pequeño seco en la boca del matraz a modo de refrigerante de reflujo. Colocar el matraz en el baño como se indica en 12.2.4. Al cabo de una hora retirar el matraz del baño y dejar enfriar.
Agregar por el embudo 1 ml de agua destilada. Si la adición del agua produce turbidez, hacerla desaparecer agregando un poco de piridina. Agitar. La mezcla se calentará por la transformación del anhídrido acético en ácido acético. Con el fin de completar esta reacción y descomponer los anhídridos grasos y mixtos que hubieran podido formarse, colocar de nuevo el matraz en el baño de glicerina durante 10 minutos. Retirar el matraz del baño, y dejar enfriar a la temperatura ambiente.
Agregar, lavando las paredes del embudo y el cuello del matraz, 5 ml de etanol de 95 por 100 v/v, conteniendo 2 ó 3 gotas de solución de fenoltaleína previamente neutralizada. Valorar con solución etanólica de hidróxido potásico 0,5 N.
Realizar simultáneamente un ensayo en blanco, en idénticas condiciones.
Cuando la fuerte coloración del medio haga difícil la observación del viraje de la fenolftaleína, utilizar como indicador una solución de azul alcalino 6B.
12.5 Cálculo.

V = volumen en ml de solución etanólica de hidróxido potásico 0,5 N de normalidad conocida, utilizados para el ensayo de la muestra.
V' = volumen en ml de solución etanólica de hidróxido potásico 0,5 N de normalidad conocida, utilizados para el ensayo en blanco.
N = normalidad exacta de esta solución.
P = peso en gramos de la muestra de grasa.
El índice de acidez se determina como en 10.
12.8 Referencias.
1. Cook, L. W. J. Am. Chem. Soc. 1922.44.392.
2. American Oil Chemists' Society. Official and Tentative Methods Cd-4-40.
3. Instituto de Racionalización del Trabajo. Una norma española 55.014.
13. PREPARACIÓN DE ÁCIDOS GRASOS INSOLUBLES
13.1 Principio.
Se entiende por ácidos grasos insolubles de una grasa, los obtenidos por saponificación de aquélla, descomposición del jabón formado y aislamiento según el procedimiento descrito posteriormente. Este debe ser aplicado rigurosamente por la posible presencia de ácidos grasos más o menos solubles en agua.
13.2 Material y aparatos.
13.2.1 Cápsula de fondo redondo de aproximadamente 1.500 mililitros.
13.3 Reactivos.
13.3.1 Solución etanólica de hidróxido potásico. Disolver 18 g. de KOH en 20 ml de agua destilada, y diluir con 50 ml de etanol de 95°.
13.3.2 Ácido sulfúrico diluido. Diluir un volumen de ácido concentrado (d = 1,84) con cuatro volúmenes de agua destilada.
13.3.3 Solución acuosa de CINa al 10 por 100 p/v.
13.4 Procedimiento.
Pesar en la cápsula de 1.500 ml aproximadamente 50 g de grasa previamente homogeneizada. Fundir lenta y progresivamente calentando hasta 115-118 °C. Agitando y frotando constantemente con una espátula metálica se añade la solución de hidróxido potásico preparada. La relación de grasa a solución etanólica de hidróxido potásico debe ser de 3 : 1.
Se mueve y frota constantemente la masa contenida en la cápsula, que se sigue calentando a fuego lento hasta que el jabón obtenido esté bastante seco para formar fragmentos que no se adhieran a la espátula por simple presión.
Verter sobre el jabón un litro de agua destilada hirviente. Mantener la ebullición de la solución jabonosa durante 45 minutos de manera que se elimine el alcohol y se obtenga una solución clara. Suprimir el calentamiento. Reemplazar el agua evaporada con agua fría. Verter con precaución 70 ml de ácido sulfúrico diluido. Evitar que trazas de jabón no descompuesto se adhieran a las paredes o al borde de la cápsula.
Llevar a ebullición y mantenerla hasta que los ácidos grasos liberados floten en forma de capa límpida (en el caso particular en que la materia grasa contenga glicéridos del ácido láurico, esta operación se hará sobre baño de agua hirviente y no con ebullición de la capa acuosa).
Lavar los ácidos dos veces con 500 ml de solución de ClNa hirviente. Después de cada lavado eliminar tan completamente como sea posible la capa acuosa. Pasar los ácidos grasos a una cápsula, adicionar sulfato sódico anhidro y filtrar sobre filtro seco.
13.5 Observaciones.
Si la preparación de los ácidos grasos tiene por objeto la determinación del peso molecular medio, asegurarse que la última capa acuosa eliminada es neutra al naranja de metilo.
Si la preparación de los ácidos grasos tiene por objeto la determinación del título, dejar los ácidos grasos cristalizar en un desecador a la temperatura del laboratorio durante 24 horas antes de la determinación.
13.6 Referencia.
1. Instituto de Racionalización del Trabajo. Una norma española 55.006.
14. TÍTULO
(Método de Dalican)
14.1 Principio.
El título es el punto de solidificación de los ácidos grasos insolubles, preparados según 13.
14.2 Material y aparatos.
14.2.1 Tubo de ensayo de vidrio de aproximadamente 27,5 milímetros de diámetro interior y 120 mm de longitud, introducido en una arandela de goma que le soporta y actúa de cierre de un frasco de boca ancha de 100 mm de diámetro exterior y 130 mm de altura. El tubo introducido en el frasco debe sobresalir su parte superior 30 mm sobre la arandela.
14.2.2 Termómetro de precisión, graduado en 1/10 ó 1/5 de grado, hasta 70 °C, aproximadamente. El depósito del termómetro tiene 20 mm de largo y 6 mm de diámetro.
14.2.3 Soporte para suspender verticalmente el termómetro en el eje del tubo de ensayo.
14.3 Procedimiento.
Situar el frasco o recipiente exterior a una temperatura de 20° ó 25 °C inferior al título presumible, sumergiéndolo exteriormente en un baño de agua caliente o fría.
Fundir los ácidos grasos llevándolos a una temperatura 10 °C aproximadamente superior al título presumible.
Verterlos en el tubo de ensayo hasta una altura de 55 milímetros, aproximadamente.
Colocar el termómetro suspendido con la mayor exactitud posible en el eje del tubo de ensayo y su extremidad inferior a 1 cm del fondo del tubo. Observar la columna de mercurio; en principio baja rápidamente y después cada vez más lentamente. Simultáneamente, se observa una cristalización de los ácidos grasos que parte del fondo del tubo y va recubriendo, poco a poco, la parte inferior del depósito del termómetro. Cuando la columna de mercurio parece pararse, después de cuatro observaciones, con cinco segundos de intervalo, inmediatamente se imprime al termómetro un movimiento circular, tres veces a la derecha y tres veces a la izquierda, teniendo cuidado de romper bien los cristales que se forman en el tubo.
Volver a colocar rápidamente el termómetro en el eje del tubo y observar de nuevo.
La columna de mercurio, después de haber bajado bruscamente durante la agitación, vuelve a subir, alcanzando un máximo, para descender de nuevo.
Este máximo se toma como punto de solidificación de los ácidos grasos o títulos.
Se realizan determinaciones hasta obtener valores concordantes, que no difieran en más de 0,2 °C.
14.4 Expresión de resultados.
En grados centígrados, indicando el método seguido.
14.5 Referencias.
1. American Oil Chemists' Society. Official and Tentativa Methods. Cc, 12-41.
2. Instituto de Racionalización del Trabajo. Una norma española 30.308.
15. ÍNDICE DE ÁCIDOS VOLÁTILES SOLUBLES E INSOLUBLES
15.1 Principio.
Se entiende por índice de ácidos volátiles solubles (o índice de Reichert-Meissl-Wollny) al número de ml de solución alcalina 0,1 N necesarios para la neutralización de los ácidos grasos volátiles solubles en agua obtenidos en las condiciones del método a partir de 5 g de grasa.
Se entiende por índice de ácidos volátiles insolubles (o índice de Polenske) al número de ml de solución alcalina 0,1 N necesarios para la neutralización de los ácidos grasos insolubles en agua, obtenidos en las condiciones del método, a partir de 5 g de materia rasa.
El método tiene carácter convencional; por tanto, exige observar rigurosamente las condiciones, prescritas, a fin de obtener resultados reproductibles.
15.2 Material y aparatos.
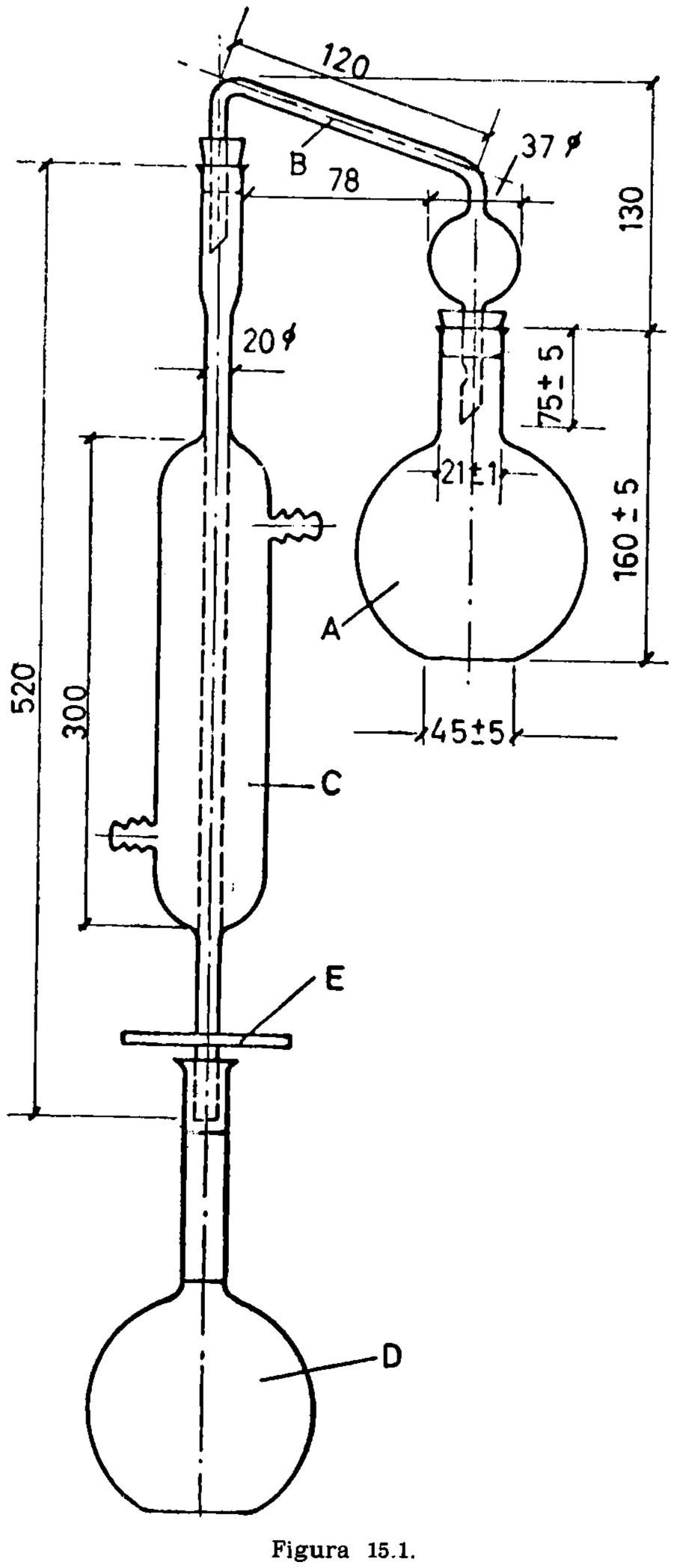
15.2.1 Aparato de vidrio, según la figura 15.1, compuesto esencialmente de las partes siguientes:
15.2.1.1 Matraz de fondo plano, de aproximadamente 300 mililitros (A).
15.2.1.2 Tubo de doble codo, adaptable a matraz y refrigerante (B).
15.2.1.3 Refrigerante (C).
15.2.1.4 Matraz de fondo plano, con tapón esmerilado, marcados en su cuello los enrases de 100 y 110 ml, tarado (D).
15.2.1.5 Placa de amianto o similar, para proteger la periferia del matraz del contacto directo de la llama (E). Dimensiones: Diámetro 120 mm (± 5 mm). Espesor, 6 mm. Orificio central circular de 45 mm de diámetro, que soporta el matraz durante el calentamiento.
15.2.2 Fuente de calor, que permite efectuar sin modificación de régimen la destilación en el tiempo prescrito.
15.3 Reactivos.
15.3.1 Glicerina (d = 1,26), correspondiente a 98 por 100, p/p, de glicerol neutro a la fenolftaleína.
15.3.2 Solución acuosa de hidróxido sódico, 44 por 100 p/p.
Eventualmente dejar calentar o filtrar al abrigo del CO2. La solución empleada debe estar perfectamente limpia y clara.
15.3.3 Agua destilada, exenta de CO2 por reciente ebullición.
15.3.4 Solución de ácido sulfúrico N.
15.3.5 Solución acuosa de hidróxido sódico o potásico, 0,1 N, de normalidad conocida exactamente.
15.3.6 Piedra pómez en granos. Tamaño de grano, entre 1,5 y 2 mm.
15.4 Procedimiento.
15.4.1 Ácidos volátiles solubles. Pesar exactamente en el matraz tarado 5 g de materia grasa a la 0,01 de gramo. Agregar 20 g (aproximadamente 16 mi) de glicerol. Agregar 2 ml de la solución de hidróxido sódico al 44 por 100 p/p, utilizando una bureta protegida contra el CO2, descartando las primeras gotas. Calentar el matraz sobre una llama libre, agitando continuamente hasta que la grasa sea completamente saponificada, en este punto el líquido no formará más espuma, y su aspecto será limpio. Se evitará todo sobrecalentamiento durante la saponificación.
Dejar enfriar hasta aproximadamente 90 °C. Agregar 93 ml de agua destilada caliente. Homogeneizar por agitación; el líquido estará limpio. Agregar de 0,6 a 0,7 g de piedra pómez en granos, después 50 ml de la solución acuosa de ácido sulfúrico N. Montar el matraz en el aparato. Calentar muy moderadamente hasta que los ácidos grasos liberados se reúnan en la superficie, formando una capa limpia. Establecer el régimen de calefacción de forma que en 19 a 21 minutos se recojan 110 ml de destilado. El destilado se recoge en el matraz aforado. La temperatura del agua a la salida del refrigerante debe estar comprendida entre 15 °C y 20 °C. La destilación comienza cuando se forma la primera gota en la extremidad del tubo acodado unida al refrigerante.
Interrumpir la calefacción cuando el destilado alcance, exactamente, el enrase de 110 ml en el matraz. Retirar rápidamente éste y colocar una probeta pequeña en su lugar.
Colocar el matraz en un termostato regulado a 15 °C, durante 10 minutos; el enrase de 110 debe estar un centímetro por debajo del nivel del líquido termostático.
Cerrar el matraz con el tapón esmerilado y homogeneizar el destilado invirtiendo 4 ó 5 veces, con suavidad, para evitar la formación de espuma. Filtrar el líquido sobre filtro sin pliegues (diámetro 8 cm). El filtrado ha de ser totalmente limpio. Medir 100 ml. Agregar 5 gotas de solución de fenolftaleína y valorar con solución alcalina 0,1 N.
Simultáneamente realizar un ensayo en blanco sin materia grasa, sustituyendo la saponificación sobre llama libre, por un calentamiento en baño de agua hirviente durante 15 minutos.
15.4.2 Ácidos volátiles insolubles. Después de la valoración de los ácidos volátiles solubles, lavar el refrigerante, el matraz aforado y la probeta pequeña, tres veces sucesivas, empleando cada vez 15 ml de agua destilada, exenta de carbónico y fría (15 °C). Las tres porciones de 15 ml se emplean para lavar, a continuación, tres veces el filtro, llenando éste hasta el borde. El último lavado recogido aparte debe ser neutralizado por una gota de solución alcalina 0,1 N.
Lavar el refrigerante, matraz aforado, probeta y filtro, tres veces sucesivas, empleando en cada operación 15 ml de etanol neutralizado. Se reúnen los líquidos alcohólicos de lavado (unos 45 ml), y se valoran con solución alcalina 0,1 N, utilizando una gota de disolución alcohólica de fenolftaleína.
15.5 Cálculo.
Índice de ácidos volátiles solubles o de Reichert-Meissl.

Índice de ácidos volátiles insolubles, o índice Polenske.

V = volumen en ml de solución alcalina 0,1 N, utilizados para el ensayo con la materia grasa.
V' = volumen en ml de solución alcalina 0,1 N, utilizados para el ensayo en blanco.
V'' -= volumen en ml de solución alcalina 0,1 N, utilizados para valorar los líquidos alcohólicos de lavado.
P = peso en g de la muestra ensayada.
N = normalidad exacta de la solución alcalina 0,1 N.
15.6 Observaciones.
Si la cantidad de muestra de que se dispone no pemite pesar 5 g, se puede realizar el análisis con una cantidad menor, siempre que se mantenga por encima de 2,5 g. En estos casos se completarán los 5 g con un aceite que tenga índices de ácidos volátiles solubles e insolubles inferior a 0,5. Se efectuará en los cálculos las correspondientes correcciones.
15.7 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. D.9.
2. American Oil Chemists' Society. Official and Tentative Methods Cd 5-40.
18(a). ÍNDICE DE IODO
(Método de Wijs)
16(a).1 Principio.
El índice de iodo de un cuerpo graso es función de su grado de insaturación. Se determina añadiendo a la muestra un exceso de reactivo halogenado, valorando el reactivo que no reacciona.
Se expresa convencionalmente por el peso de iodo absorbido por cien partes en peso de la materia grasa.
16(a).2 Material y aparatos.
16(a).2.1 Navecillas de vidrio de 2 a 3 ml de capacidad.
16(a).2.2 Matraces erlenmeyer de vidrio, de boca ancha, con tapón esmerilado, de aproximadamente 300 ml.
Todo el material debe estar perfectamente limpio y seco.
16(a).3 Reactivos.
16(a).3.1 Solución acuosa de ioduro potásico al 10 por 100 p/v, Esta solución debe estar exenta de iodo y de iodato potásico.
16(a).3.2 Solución acuosa de tiosulfato sódico, 0,1 N,
16(a).3.3 Tetracloruro de carbono puro. Comprobar que está exento de materias oxidables. Agitando 10 ml con 1 ml de solución acuosa saturada de dicromato potásico y 2 ml de ácido sulfúrico concentrado, no debe aparecer coloración verde.
16(a).3.4 Engrudo de almidón.
16(a).3.5 Reactivo de Wijs.
16(a).3.5.1 Con tricloruro de iodo.
Pesar 9 g de tricloruro de iodo ICl3 en un matraz de vidrio topacio de 1.500 ml; disolver en un litro de una mezcla compuesta de 700 ml de ácido acético y 300 ml de tetracloruro de carbono.
Determinar el contenido en halógeno de la forma siguiente: Tomar 5 ml y agregar 5 ml de la solución acuosa de ioduro potásico y 30 ml de agua. Valorar con solución de tiosulfato sódico, 0,1 N en presencia de engrudo de almidón como indicador.
Agregar al reactivo 10 g de iodo pulverizado, y disolver agitando.
Determinar el contenido en halógeno como anteriormente; debe ser igual a vez y media de la primera determinación. Agregar todavía una pequeña cantidad de iodo, de forma que sobrepase ligeramente el límite de vez y media, por ser necesario que no quede ninguna traza de tricloruro de iodo, cuya presencia provocaría reacciones secundarias.
Dejar decantar después de verter el líquido claro en un matraz o frasco de color topacio La solución, bien conservada al abrigo de la luz, puede ser utilizada durante varios meses.
16(a).3.5.2 Con monocloruro de iodo.
Disolver 19 g de monocloruro de iodo en 1 litro de una mezcla de 700 ml de ácido acético y 30 ºC ml de tetracloruro de carbono. Después de agregar una pequeña cantidad de iodo puro (algunos miligramos), determinar el contenido en halógeno, como se realizó anteriormente, y diluir si es necesario con la mezcla de disolventes hasta que 5 ml de reactivo correspondan aproximadamente a 10 ml de solución de tiosulfato sódico 0,1 N.
16(a).4 Procedimiento.
Según el índice de iodo previsto, la toma de muestra variará de la forma siguiente:
| índice de iodo previsto |
Toma de maestra g |
|---|---|
| < 5 | 3,00 |
| 5 a 20 | 1,00 |
| 21 a 50 | 0,60 |
| 51 a 100 | 0,30 |
| 101 a 150 | 0,20 |
| 151 a 200 | 0,15 |
En una pequeña navecilla de vidrio, pesar exactamente la cantidad necesaria con una aproximación de 1 mg. Introducir la navecilla y su contenido en un erlenmeyer con tapón esmerilado de aproximadamente 300 ml. Agregar 15 ml de tetracloruro de carbono y disolver. Agregar exactamente 25 ml del reactivo. Tapar el matraz, agitar ligeramente y protegerlo de la luz.
Dejar estar 1 hora para grasas cuyo índice sea inferior a 150 y 2 horas para las de índice superior a 150 y los aceites polimerizados u oxidados.
Agregar 20 ml de la solución de ioduro potásico y 150 ml de agua.
Valorar con solución de tiosulfato sódico 0,1 N, con engrudo de almidón como indicador, hasta desaparición justa del color azul después de agitación intensa.
Hacer un ensayo en blanco sin materia grasa en las mismas condiciones.
16(a).5 Cálculo.

P = peso en g de la muestra.
V = volumen en ml de la solución de tiosulfato sódico 0,1 N utilizados para el ensayo en blanco.
V' = volumen en ml de solución de tiosulfato sódico 0,1 N utilizados para la materia grasa.
N = normalidad de la solución de tiosulfato sódico utilizada.
16(a).6 Referencias.
1. Z. angew Chem 1898. 11, 291.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.013.
16(b) ÍNDICE DE IODO
(Método de Hanus)
16(b).1 Principio.
Como 16(a).1.
16(b).2 Material y aparatos.
16(b).2.1 Frasco de boca ancha, de 200 a 220 ml de capacidad, con tapón esmerilado.
16(b).2.2 Buretas graduadas de 0,1 de ml, comprobadas y contrastadas.
16(b).2.3 Buretas o pipetas de 25 ml de salida rápida.
16(b).3 Reactivos.
16(b).3.1 Tetracloruro de carbono, inerte a la solución de Hanus.
16(b).3.2 Solución de ioduro potásico al 10 por 100, exenta de iodo y de iodatos.
16(b).3.3 Solución de tiosulfato sódico 0,1 N.
16(b).3.4 Engrudo de almidón.
16(b).3.5 Reactivo de Hanus: Disolver en un frasco de color amarillo, con tapón esmerilado, 10 g de monobromuro de iodo en 500 ml de ácido acético cristalizable (99-100 por 100), exento de etanol.
16(b).4 Procedimiento.
Trabajar en luz difusa.
En el frasco limpio y seco, pesar de 0,25 a 0,30 g de materia grasa limpia y filtrada sobre papel (tomar una cantidad de grasa tal que, una vez finalizada la reacción, deberá quedar sin absorber una cantidad de iodo igual, por lo menos, al 70 por 100 de la cantidad total de iodo añadida.
Disolver la materia grasa en 10 ml de tetracloruro de carbono. Añadir, mediante bureta o pipeta de salida rápida, 25 mililitros exactos del reactivo de Hanus.
Tapar el frasco, mezclar por agitación suave. Dejar reposar al abrigo de la luz durante una hora, a una temperatura de 20°± 5 °C.
Añadir 20 ml de solución de ioduro potásico, y 100 ml de agua destilada. Mezclar.
Valorar con la solución de tiosulfato sódico 0,1 N (utilizando engrudo de almidón como indicador), agitando constantemente. Añadir el engrudo de almidón poco antes de finalizar la valoración.
Realizar una prueba en blanco en idénticas condiciones.
16 (b).5 Cálculo.

V = volumen en ml de solución de tiosulfato sódico 0,1 N utilizados en la prueba en blanco.
V' = volumen en ml de solución de tiosulfato sódico 0,1 N utilizados en el ensayo con la muestra.
P = peso en g de la muestra utilizados en la determinación.
16 (b).6 Referencias.
1. Ztschr. Untersuch. Nahr. und Genussm. 1901. 4, 913, 20.
2. Consejo Oleícola Internacional, 1967.
3. Instituto de Racionalización del Trabajo. Una Norma Española 55.013.
17. ÍNDICE DE TIOCIANÓGENO
17.1 Principio.
El índice de tiocianógeno de una materia grasa es función de su grado de insaturación. En la práctica se determina por la fijación de tiocianógeno, y convencionalmente se expresa por el peso de iodo equivalente al tiocianógeno absorbido por 100 partes en peso de la grasa.
El tiocianógeno (SCN)2 se fija sobre los dobles enlaces como los halógenos. Es menos reactivo que los halógenos y su fijación sólo es cuantitativa con el ácido oleico
Se considera que aproximadamente el (SCN)2 se fija sobre un doble enlace del ácido linoleico y sobre dos dobles enlaces del ácido linolénico.
17.2 Material y aparatos.
17.2.1 Navecillas de vidrio de 2 a 3 ml.
17.2.2 Matraces de vidrio de boca esmerilada.
17.2.3 Embudo Büchner.
Todo el material debe estar perfectamente lavado y seco.
17.3 Reactivos.
17.3.1 Ácido acético glacial puro. Antes de su empleo calentarle a ebullición con refrigerante de reflujo, durante 3 horas, con 10 por 100 v/v de anhídrido acético. Destilar sobre anhídrido fosfórico.
17.3.2 Tiocianato de plomo. Disolver 250 g de acetato de plomo, químicamente puro (CH3COO)2 Pb, 3H2O en 500 ml de agua destilada; disolver 250 g de tiocianato potásico en 500 mililitros de agua destilada, mezclar las dos soluciones, filtrar el tiocianato de plomo precipitado sobre embudo Büchner. Lavar con agua destilada, después con etanol de 95 por 100 v/v, y por último con óxido de etilo. Secar el precipitado tan completamente como sea posible, mediante aspiración por vacío; recoger el precipitado en una cápsula de porcelana, y colocar ésta en un desecador con anhídrido fosfórico, durante ocho o diez días. El tiocianato de plomo así obtenido debe presentar color débilmente amarillo blanquecino o verduzco. Si se presenta coloración más intensa, volver a someterle al mismo tratamiento. Guardando en frasco amarillo-pardo, al abrigo de la luz, puede conservarse hasta dos meses. Si se ha coloreado intensamente no utilizarlo.
17.3.3 Bromo puro, previamente desecado.
17.3.4 Tetracloruro de carbono; agitar dos veces el tetracloruro de carbono empleando cada vez el 5 por 100 v/v de ácido sulfúrico concentrado. Separar el tetracloruro de carbono. Lavar con agua destilada, después con 5 por 100 v/v de una solución acuosa de hidróxido potásico al 50 por 100 p/v. Agitar con hidróxido potásico en pastillas. Destilar sobre anhídrido fosfórico.
17.3.5 Ioduro potásico exento de iodo y iodato.
17.3.6 Solución acuosa de tiosulfato sódico 0,1 N de normalidad conocida.
17.3.7 Solución de tiocianógeno aproximadamente 0,2 N. Poner en suspensión 50 g de tiocianato de plomo en 500 ml de ácido acético. Disolver, por separado, 5,1 ml de bromo en 100 ml de ácido acético. Verter por pequeñas porciones la solución de bromo en la solución de tiocianato de plomo, agitando vigorosamente después de cada adición hasta decoloración completa. Dejar decantar. Filtrar tan rápidamente como sea posible por embudo Büchner, previamente secado, recogiendo el filtrado en un matraz seco.
Colocar el embudo Büchner sobre un segundo matraz y filtrar de nuevo; el filtrado debe ser limpio e incoloro. Conservarle en un frasco de vidrio de color amarillo o pardo, de tapón esmerilado, perfectamente seco, y en lugar fresco (temperatura inferior a 20 °C). De no observar estas condiciones se altera rápidamente. Antes de su utilización, comprobar que permanece incoloro.
17.4 Procedimiento.
Pesar con una precisión de 1 mg, en una navecilla de vidrio, la cantidad necesaria de materia grasa, según las indicaciones de la tabla siguiente (la cantidad de solución de tiocianógeno será de un 150 por 100 en exceso):
| Índice previsto |
Peso de la muestra g |
Reactivo ml |
|---|---|---|
| 0 a 30 | ≅ 0,50 | 25 |
| 30 a 50 | ≅ 0,30 | 25 |
| 50 a 100 | ≅ 0,25 | 25 |
| 100 a 150 | ≅ 0,20 | 50 |
| Superior a 150 | ≅ 0,15 | 50 |
Introducir la navecilla y su contenido en un matraz de boca esmerilada. Agregar 10 ml de tetracloruro de carbono. Disolver. Agregar 25 ó 50 ml exactamente medidos de la solución de tiocianógeno. Tapar el matraz. Mezclar por agitación. Dejar en la oscuridad durante 24 horas, a una temperatura de aproximadamente 20 °C.
Agregar 2 g de ioduro potásico pulverizado. Agitar durante 3 minutos. Agregar 30 ml de agua destilada. Valorar en presencia de engrudo de almidón, con la solución de tiosulfato sódico 0,1 N, hasta desaparición del color azul después de enérgica agitación.
Realizar un ensayo en blanco, sin materia grasa, en las mismas condiciones.
La solución de tiocianógeno debe valorarse antes de iniciar los ensayos. Si la diferencia entre esta valoración y la del ensayo en blanco es superior 0,2 ml, significa que la solución se descompone muy rápidamente; repetir las operaciones con otra solución más correcta.
Los ensayos sobre materia grasa y en blanco se realizarán al menos por duplicado. Si entre ambos ensayos, sobre materia grasa y en blanco, se obtienen diferencias superiores a 2 unidades del índice de tiocianógeno, repetir la determinación.
17.5 Cálculo.

V = volumen en ml de solución de tiosulfato sódico 0,1 N utilizados en el ensayo en blanco.
V' = volumen en ml de solución de tiosulfato sódico 0,1 N utilizados en un ensayo con materia grasa.
P = peso en g de la muestra ensayada.
N = normalidad exacta de la solución de tiosulfato sódico 0,1 N.
17.6 Referencias.
1. H. P. Kaufmann, Chem. Ztg. 1925. 49, 768.
2. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps 1964. II. D.8.
18. ÍNDICE DE POLIBROMUROS
18.1 Principio.
El índice de polibromuros de una sustancia grasa es el peso en g de polibromuros obtenidos, en las condiciones descritas a partir de 100 g de grasa.
El método tiene carácter convencional, por lo que sus prescripciones han de ser observadas rigurosamente.
18.2 Material y aparatos.
18.2.1 Matraz de saponificación, de unos 200 ml, con boca esmerilada adaptable a un tubo refrigerante de reflujo de aproximadamente 1 m de largo.
18.2.2 Matraz aforado de 100 ml.
18.2.3 Matraz erlenmeyer de unos 100 ml de capacidad.
18.2.4 Ampollas de decantación de 500 ml.
18.2.5 Crisol con placa filtrante G3, con dispositivo para refrigerar y kitasato para recoger el filtrado.
18.3 Reactivos.
18.3.1 Disolución alcohólica de hidróxido potásico 1 N.
18.3.2 Disolución de ácido clorhídrico 1 N.
18.3.3 Disolución acuosa de anaranjado de metilo al 0,1 por 100.
18.3.4 Éter etílico, seco.
18.3.5 Solución acuosa al 10 por 100 de cloruro sódico.
18.3.6 Sulfato sódico anhidro.
18.3.7 Disolución de bromo. Agregar 4 ml de bromo puro y seco, enfriados a 0 °C, a 100 ml de éter etílico seco, previamente enfriados a la misma temperatura. Realizar la adición de bromo por pequeñas porciones, dejándolas resbalar por las paredes del recipiente y agitando en cada adición. Mantener a 0° hasta el momento de su utilización.
18.4 Procedimiento.
18.4.1 Preparación de los ácidos grasos. Pesar con precisión de 10 mg, 5 g de grasa en el matraz de saponificación. Agregar 25 ml de potasa alcohólica 1 N. Acoplar el refrigerante y calentar a ebullición, con una pequeña llama, durante 30 minutos.
Al principio de la calefacción agitar el matraz por rotación suave, para homogeneizar su contenido, a fin de evitar el sobrecalentamiento de la grasa. Terminada la saponificación, separar el refrigerante. Agregar 50 ml de agua destilada y pasar el contenido del matraz a la ampolla de decantación, utilizando en varias veces, 80 ml de agua en total. Agregar 50 ml de éter etílico y ligero exceso de ácido clorhídrico 1 N, calculado mediante la adición de dos gotas de anaranjado de metilo. Agitar fuertemente la ampolla para disolver totalmente los ácidos grasos liberados. Dejar reposar hasta que se separen netamente las dos capas. Pasar la disolución hidroalcohólica a otra ampolla de decantación y extraer nuevamente con otros 50 ml de éter etílico, repitiendo la operación anterior. Reunir los dos extractos etéreos en una misma ampolla y lavar tres veces, con 50 ml cada una, con la disolución de cloruro sódico. En los tres lavados se obtendrá una buena decantación, por adecuada separación de las dos capas.
Pasar la disolución etérea a un matraz aforado de 100 ml y completar hasta el enrase con éter etílico. Añadir una pequeña cantidad de sulfato sódico anhidro (de 1 a 2 g) para asegurar la deshidratación. Tapar el matraz y agitar para homogeneizar.
En el caso de que la bromación no vaya a realizarse inmediatamente, llenar el matraz con CO2 o con N2, tapar y conservar en lugar oscuro.
18.4.2 Bromuración. Pesar el matraz erlenmeyer de 100 mililitros, previamente secado en estufa y enfriado en desecador. Agregar una cantidad de polibromuros del orden de 0,1 g, obtenidos a partir del mismo aceite u otro de naturaleza análoga; pesar nuevamente el matraz. Los polibromuros han de estar perfectamente divididos.
Agregar al matraz 20 ml, exactamente medidos con pipeta, de la disolución etérea de los ácidos grasos. Agitar para asegurar la disolución de los polibromuros. Tapar el matraz e introducirlo en hielo fundente durante 15 minutos, agitando de cuando en cuando, para saturar el líquido de polibromuros.
Añadir 20 ml de la disolución de bromo, poco a poco y resbalando por la pared, agitar en cada adición, cuidando de que la temperatura no exceda nunca de + 2 °C. Una vez que se han agregado los dos tercios del reactivo, se puede añadir el resto más rápidamente, utilizando las últimas porciones para limpiar las paredes del matraz. Tapar y dejar en hielo fundente durante 3 horas,
Realizar un ensayo en blanco, en las mismas condiciones.
18.4.3 Filtración y lavados. Al cabo de las 3 horas, filtrar la disolución de los ácidos grasos por el crisol de placa filtrante, previamente desecado en estufa a 105°, enfriado en desecador y tarado. El crisol estará rodeado de hielo fundente; facilitar 1 filtración mediante un débil vacío, que será eliminado antes de que se consuma el líquido sobre la placa filtrante.
Lavar cuatro veces la torta de polibromuros con disolución de polibromuros en éter etílico (0,06 g en 100 ml), enfriada a 0 °C. En los dos primeros lavados utilizar 20 ml cada vez, y en los dos últimos, 10 ml cada vez. Adicionar estos volúmenes de éter sucesivamente al matraz donde se ha efectuado la precipitación, cuidando de lavar bien las paredes y agitando enérgicamente para desprender las partículas adheridas. Después de efectuar estas operaciones conviene introducir el matraz en hielo fundente, y no verter el éter en el crisol hasta asegurarse de haberlo enfriado a 0 °C. Cuando se opera con aceite de índice de polibromuros bajo pueden reducirse los lavados a tres solamente, utilizando en cada uno un volumen de 10 ml.
18.4.4 Desecación y pesada. Colocar en estufa los crisoles y erlenmeyer que se han utilizado en la precipitación; elevar progresivamente la temperatura hasta alcanzar de 95° a 100 °C. Dejar 30 minutos a esta temperatura; enfriar en desecador y pesar.
18.5 Cálculo.

P' = aumento de peso en g del conjunto matraz y crisol, empleados en el ensayo de la muestra.
P" = pérdida de peso en g del conjunto crisol y matraz, empleados en el ensayo en blanco.
P = peso en g de la muestra.
18.6 Referencia.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. D.11.
19. ÍNDICE DE DIENOS
19.1 Principio.
El índice de dienos representa la cantidad de iodo expresada en g equivalente al anhídrido maleico, fijado por los dobles enlaces presentes en 100 g de grasa.
19.2 Material y aparatos.
19.2.1 Matraz de 250 ml de boca esmerilada y refrigerante de reflujo.
19.2.2 Baño de aceite o manta eléctrica.
19.2.3 Pipeta de 25 ml.
19.2.4 Embudo de separación de 250 ml.
19.2.5 Matraz de 300 ml.
19.3 Reactivos.
19.3.1 Solución al 6 por 100 de anhídrido maleico (p.f. 52-54 °C), en toluol puro; dejar en reposo 24 horas. Filtrar con papel de filtrado rápido antes de su uso.
19.3.2 Disolución de hidróxido sódico N.
19.3.3 Éter etílico.
19.4 Procedimiento.
Filtrar la grasa a través de un filtro seco.
Pesar con precisión de 1 mg aproximadamente 3 g de materia grasa en el matraz de 250 ml, agregar 25 ml de solución de anhídrido maleico, hervir moderadamente a reflujo durante 3 horas, dejar enfriar unos minutos, agregar a través del refrigerante 5 ml de agua destilada, y volver a hervir durante 15 minutos. Dejar enfriar a la temperatura ambiente, agregar a través del refrigerante 5 ml de éter etílico, y después 20 ml de agua destilada. Quitar el refrigerante y pasar el contenido del matraz al embudo de separación. Lavar el matraz tres veces, con 7 ml de éter etílico cada vez, y después otras tres veces, con 8 ml de agua destilada cada vez, incorporando todos los lavados al embudo de separación. Agitar y dejar reposar hasta la separación de las dos capas. Pasar la capa acuosa al matraz erlenmeyer de 300 ml. Repetir la extracción del extracto etéreo contenido en el embudo de separación, con 25 ml y después con 10 ml de agua destilada, incorporar la capa acuosa al matraz de 300 ml.
Valorar todos los extractos acuosos reunidos con disolución de hidróxido sódico N, usando fenolftaleína como indicador. Realizar simultáneamente un ensayo en blanco.
19.5 Cálculo.

V = volumen en ml de NaOH empleados en el ensayo en blanco.
V' = volumen en ml de NaOH empleados en el ensayo de la grasa.
N = normalidad en la solución de hidróxido sódico.
P = peso en g de la materia grasa.
19.6 Observaciones.
Es conveniente que el agua empleada sea destilada, recientemente hervida y enfriada.
19.7 Referencias.
1. H. P. Kaufmann, J. Baltés. Fette und Seifen 1936. 43, 93.
2. American Oil Chemists' Society. Official and Tentative Methods. Ka 12-55.
20. ÁCIDOS OXIDADOS
20.1 Principio.
Este método determina los ácidos oxidados contenidos en las grasas, expresados en materia grasa insoluble en éter de petróleo.
Teniendo carácter empírico, exige observar estrictamente las condiciones prescritas.
20.2 Material y aparatos.
20.2.1 Matraz de 150 ml, adaptable a refrigerante de reflujo.
20.2.2 Ampollas de decantación de 500 ml.
20.2.3 Estufa estabilizada a 103 °C (± 2 ºC).
20.2.4 Crisol de porcelana de 45 mm de altura y 40 mm de diámetro.
20.3 Reactivos.
20.3.1 Solución atanólica de hidróxido potásico, aproximadamente 2 N. Disolver 112 g de potasa en 500 ml de etanol de 96°, y una vez fría la solución, enrasar a 1.000 ml con etanol de 96°.
20.3.2 Éter de petróleo (p. e. 40°- 60 °C) bidestilado, exento de residuo.
20.3.3 Solución de ácido clorhídrico N.
20.3.4 Éter etílico (C2H5)2O, d = 0,712 - 0,714.
20.3.5 Etanol, 95° exento de acidez.
20.4 Procedimiento.
Tratar 5 g de grasa pesados con una aproximación de 10 miligramos como en 22(b).4, hasta agotamiento de la disolución hidroalcohólica de jabón por el éter y los dos primeros lavados.
Colocar la disolución hidroalcohólica y el agua procedente de los dos primeros lavados del éter en una cápsula de 500 mililitros. Hervir hasta que queden eliminadas las trazas de éter disuelto y el alcohol en su totalidad.
Trasvasar el contenido de la cápsula a una ampolla de decantación de 500 ml, lavando la cápsula con pequeñas cantidades de agua, procurando que el volumen total del líquido en la ampolla sea de 150 ml aproximadamente.
Añadir solución de ácido clorhídrico N en ligero exceso (51 ml aproximadamente). Agitar 2 minutos. Después de agitado no ha de quedar espuma; en caso contrario se añade un poco más de ácido.
Agregar a la ampolla 100 ml de éter de petróleo. Agitar 1 minuto. Dejar reposar 12 horas. Decantar el agua ácida. Filtrar la solución de éter sobre un filtro lento de 90 mm de diámetro.
Los ácidos oxidados se adhieren generalmente a las paredes del tubo y aparecen en forma de una masa roja oscura.
Si los ácidos oxidados son abundantes, hacer salir el éter de petróleo por la parte superior de la ampolla, para evitar que los ácidos obturen la llave de salida. Lavar dos veces la ampolla de decantación con 25 ml de éter de petróleo y filtrar. Lavar también con éter de petróleo el tubo de vaciado de la ampolla y el embudo, el filtrado y el pico del embudo con porciones de 10, 10 y 5 ml respectivamente. Seguidamente desecar el exterior del pico del embudo y el del tubo de vaciado del embudo de decantación.
Disolver en etanol de 95° por 100 v/v caliente, los ácidos oxidados contenidos en la ampolla: Una vez con 25 ml de etanol caliente, y otra con 50 ml de etanol caliente. Pasar sucesivamente cada porción caliente por el filtro. Lavar el tubo de vaciado de la ampolla, el filtro y el cono del embudo con porciones respectivas de 5 ml de etanol caliente, recoger las soluciones alcohólicas en un vaso de 400 ml.
Evaporar el etanol hasta un volumen muy pequeño (algunos ml).
Transvasar cuantitativamente el residuo a un crisol de porcelana tarado, utilizando pequeñas porciones de éter etílico (así se evitan las dificultades presentadas por la evaporación del alcohol, teniendo este disolvente tendencia a subir por las paredes del crisol).
Comenzar la evaporación al aire libre. Terminarla en baño de agua hirviente, hasta la desaparición de los olores de éter y alcohol y la aparición de un olor acre.
Colocar el crisol en estufa a 103 °C (± 2 ºC), durante media hora. Pasar a desecador con ácido sulfúrico durante 15 minutos y pesar. Repetir sucesivamente estas operaciones hasta pesada constante con una precisión de 1 mg. La última pesada representa el peso de los ácidos oxidados más el de las sales minerales arrastradas con los ácidos oxidados.
Calcinar el residuo, dejar enfriar el crisol y pesar.
20.5 Cálculo.

P' = peso en g de los ácidos oxidados, más las sales minerales.
P" = peso en g de las sales minerales.
P = peso en g de la muestra tomada para análisis.
20.6 Referencias.
1. International Union of Pure and Applied Chemistry Standard Methods for the Analysis of Oils. Fats and Soaps. 1964. II. D.12.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.008.
21. ÍNDICE DE PERÓXIDOS
21.1 Principio.
Se denomina «índice de peróxidos» a los miliequivalentes de oxígeno activo contenidos en un kilogramo de la materia ensayada, calculados a partir del yodo liberado del yoduro potásico, operando en las condiciones que se indican en la metódica.
Las sustancias que oxidan el yoduro potásico en las condiciones descritas se supone son peróxidos u otros productos similares de oxidación de la grasa, por lo que el índice obtenido puede tomarse, en una primera aproximación, como una expresión cuantitativa de los peróxidos de la grasa.
21.2 Material y aparatos.
21.2.1 Navecillas de vidrio de aproximadamente 3 ml para pesada de la grasa.
21.2.2 Matraces con tapón esmerilado, de aproximadamente 250 ml, previamente secados y llenos de gas inerte (anhídrido carbónico o nitrógeno).
21.3 Reactivos.
21.3.1 Cloroformo, para análisis, exento de oxígeno por barboteo de una corriente de gas inerte puro y seco.
21.3.2 Ácido acético glacial puro exento de oxígeno como en 2.3.1.
21.3.3 Solución acuosa saturada de yoduro potásico, exento de yodo y yodatos.
21.3.4 Soluciones acuosas de tiosulfato sódico 0,01 N y 0,002 N exactamente valoradas.
21.3.5 Solución indicadora de almidón al 1 por 100 en agua destilada.
21.4 Procedimiento.
Tomar un matraz con cierre esmerilado, de unos 250 ml, previamente seco, y llenar con un gas inerte, puro y seco (anhídrido carbónico o nitrógeno). Introducir tan rápidamente como se pueda la muestra del aceite que se desea ensayar, definida en función de los índices presumidos (ver 21.6.1).
Agregar 10 ml de cloroformo, en el cual se disuelve rápidamente la grasa por agitación, 150 ml de ácido acético glacial y 1 ml de una disolución acuosa de yoduro potásico.
Cerrar el matraz y mantener en agitación durante un minuto, imprimiéndole un suave movimiento de rotación, conservándolo después en la oscuridad durante 5 minutos; transcurrido este tiempo, agregar 75 ml de agua, agitar vigorosamente y valorar el yodo liberado con una disolución de tiosulfato 0,002 N, para los aceites de índices inferiores o iguales a 20 y 0,01 N para los índices más elevados.
Paralelamente, se efectúa un ensayo testigo, sin aceite, que debe dar un índice nulo.
21.5 Cálculos.
El índice de peróxidos se expresa en miliequivalentes de oxígeno por kilogramo de muestra, y se calculará aplicando la fórmula siguiente:

V = tiosulfato, en ml, consumido en la valoración.
N = normalidad de la solución de tiosulfato.
P = peso, en gramos, de la muestra de grasa tomada para la determinación.
21.6 Observaciones.
21.6.1 Peso de la muestra. La toma de las muestras para el ensayo se efectuará tomando una cantidad de grasa de acuerdo con el índice de peróxidos que se presupone y que se indica en el cuadro siguiente:
| Índice que se presupone | Peso de la muestra en g |
|---|---|
| De 0 a 20 | De 2,0 a 1,2 |
| De 20 a 30 | De 1,2 a 0,8 |
| De 30 a 50 | De 0,8 a 0,5 |
| De 50a 100 | De 0,5 a 0,3 |
21.6.2 Para la expresión del índice de peróxidos se han propuesto otras unidades distintas a la adoptada en esta norma y que suelen ser utilizadas, en algunos casos, presentándose a confusiones en la interpretación de resultados. Para evitar estos errores y los inconvenientes que pudieran derivarse de los mismos, en los informes analíticos deberá indicarse siempre la unidad en la que se expresa el índice.
Para facilitar el paso de una unidad a otra, se indican a continuación, los factores de conversión por los que deberá multiplicarse, en cada caso, la cifra del índice, expresado en una determinada unidad, para obtener la cifra equivalente en la unidad que se define en 21.1.
| Índice de peróxidos expresado en | Factor de con versión para calcular el índice expresado en miliequivalentes de oxígeno activo por kilogramo de grasa |
|---|---|
| a) Microgramos de oxígeno activo por gramo de grasa . | 0,125 |
| b) Gramos de oxígeno activo por kilogramo de grasa. | 125 |
| c) Mililitros de solución de tiosulfato sódico 0,01 N por kilogramo de grasa. | 0,01 |
| d) Mililitros de solución de tiosulfato sódico 0,01 N por gramo de grasa. | 10 |
| e) Mililitros de solución de tiosulfato sódico 0,002 N por gramo de grasa. | 2 |
| f) Milimoles de oxígeno activo por kilogramo de grasa. | 2 |
21.7 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.023.
2 International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps 1964. II. D.13.
22(a). INSAPONIFICABLE
(Método éter de petróleo)
22(a).1 Principio.
Se entiende por insaponificable el peso en g de sustancias no saponificables, insolubles en agua y solubles en el disolvente utilizado en la determinación, contenidas en 100 g de grasa.
El método es aplicable a todas las materias grasas. Su exactitud es sólo aproximada para aquellas grasas con un contenido de insaponificable muy elevado.
22(a).2 Material y aparatos.
22(a).2.1 Matraz de fondo plano, de 200 ml, adaptable a refrigerante de reflujo.
22(a).2.2 Refrigerante de reflujo.
22(a).2.3 Embudos de separación de 500 ml.
22(a).2.4 Estufa graduable a 103° 1± 2 °C).
22(a).3 Reactivos.
22(a).3.1 Solución de hidróxido potásico, alcohólica, 2 N, en etanol, de 95 por 100 v/v.
22(a).3.2 Éter de petróleo (p. e. 40° - 60°; índice de bromo, 1), redestilado y exento de residuo.
22(a).3.3 Etanol al 50 por 100 en volumen.
22(a).3.4 Agua destilada.
22(a).3.5 Solución de heliantina al 1 por 100.
22(a).3.6 Solución de ácido clorhídrico 0,1 N.
22(a).4 Procedimiento.
Eliminar el agua de la muestra por decantación y filtración sobre papel, efectuadas a una temperatura ligeramente superior al punto de fusión de determinados componentes sólidos que hubieran podido separarse de la materia grasa fluida.
Pesar en el matraz 5 g de materia grasa, con una aproximación de 0,01 g.
Añadir 50 ml de solución alcohólica de potasa 2 N. Adaptar refrigerante de reflujo. Calentar una hora con ligera ebullición. Separar la fuente de calor. Agregar por la parte superior del refrigerante 50 ml de agua destilada y agitar. Dejar enfriar.
Transvasar el contenido del matraz a un embudo de decantación, aclarar y arrastrar las últimas porciones lavando el matraz varias veces con 50 ml de éter de petróleo en total.
Agitar enérgicamente durante un minuto. Dejar en reposo hasta la completa separación de las dos fases. Pasar a un segundo embudo de decantación la fase jabonosa. Extraer ésta con otros 50 ml de éter de petróleo; decantarla de nuevo y volver a extraer con 50 ml de éter de petróleo.
Reunir las tres fracciones etéreas en un mismo embudo de separación y lavarlas tres veces seguidas, utilizando cada vez 50 ml de etanol al 50 por 100 v/v. Transvasar la solución de éter de petróleo a un pequeño matraz de cuello corto, previamente tarado y eliminar el disolvente por destilación en baño de agua hirviendo.
Secar colocando el matraz en posición horizontal en una estufa regulada a 103° durante 15 minutos. Dejar enfriar en desecador y pesar. Repetir la operación hasta que las pérdidas entre dos pesadas consecutivas sean inferiores a 0,10 por 100.
Incinerar las sustancias insaponificables obtenidas, y si dejan cenizas, dosificar su alcalinidad en presencia de heliantina con una solución de ácido clorhídrico 0,1 N (1 ml de esta solución representa 0,032 g de jabón de potasa, que hay que deducir).
22(a).5 Cálculo.
Calcular el insaponificable expresado en porcentaje.
22(a).5.1 Insaponificable

22(a).5.2 En el caso de haber incinerado el residuo y valorado con KOH alcohólica 0,1 N.

P' = peso en g del residuo.
P = peso en g de la muestra.
N = normalidad exacta de la solución acuosa de ClH 0,1 N.
V = volumen en ml de ClH 0,1 N utilizados.
22(a).6 Observaciones.
22(a).6.1 En los certificados de análisis, indicar: Método del éter de petróleo.
22(a).7 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils. Fats and Soaps. 1964. II. D.5.
2. Instituto de Racionalización del Trabajo: Una Norma Española 30.305.
22(b). INSAPONIFICABLE
(Método éter etílico)
22(b).1 Principio.
Como 22(a).1
22(b).2 Material y aparatos.
Como 22(a).2.
22(b).3 Reactivos.
22(b).3.1 Solución etanólica de hidróxido potásico, aproximadamente 2 N, en etanol de 95° v/v.
22(b).3.2 Solución acuosa de hidróxido potásico, aproximadamente 0,5 N.
22(b).3.3 Éter etílico neutro, recién destilado y exento de residuo.
22(b).4 Procedimiento.
Eliminar el agua de la muestra por decantación y filtración sobre papel, efectuadas a una temperatura ligeramente superior al punto de fusión de determinados componentes sólidos que hubieran podido separarse de la materia grasa fluida.
Pesar en el matraz, con una precisión de 0,01 g, aproximadamente, 5 g de materia grasa. Saponificar como en 22(a).4.
Separar la fuente de calor. Desconectar el refrigerante. Transvasar el contenido del matraz a una ampolla de decantación. Lavar con 100 ml de agua destilada.
Enjuagar el matraz y el refrigerante con 100 ml de éter etílico, y pasarlos a la ampolla; tapar y agitar vigorosamente, mientras el contenido esté ligeramente caliente. Dejar en reposo hasta separación nítida de las dos capas. Si aparece una emulsión persistente causada por una alcalinidad fuerte del medio, añadir unas gotas de ácido clorhídrico N.
Separar la capa alcohólico-acuosa y verterla en el matraz empleado en la saponificación.
Pasar la capa etérea a una segunda ampolla de decantación conteniendo 40 ml de agua.
Tratar la solución alcohólico-acuosa de jabón dos veces más, con porciones de 100 ml de éter etílico. Reunir las tres fracciones etéreas en la segunda ampolla de decantación. Si las fracciones etéreas reunidas contuviesen materias sólidas en suspensión, filtrar y lavar cuantitativamente el filtro con un poco de éter.
Girar, sobre sí mismo, sin sacudidas violentas, la ampolla que contiene el éter y los 40 ml de agua. Una vez separadas las dos capas, eliminar la capa acuosa. Lavar la capa etérea dos veces, con 40 ml de agua cada vez, agitando enérgicamente. Después, lavar sucesivamente con 40 ml de solución de potasa acuosa 0,5 N, con 40 ml de agua, y de nuevo con 40 ml de solución acuosa de potasa 0,5 N, y, por lo menos, dos veces con 40 ml de agua. Continuar los lavados con agua hasta que las aguas de lavado no den coloración rosa a la fenolftaleína.
Transvasar cuantitativamente la solución etérea a un matraz tarado de 200 ml, después de reducirla a pequeño volumen por evaporación. Agregar 6 ml de acetona y eliminar completamente el solvente volátil, por medio de una ligera corriente de aire, estando el matraz casi sumergido en un baño de agua hirviendo, en posición oblicua y haciéndole girar. Terminar el secado en estufa a 103 °C, como en 22(a).4.
Después de pesar el residuo, disolverlo en 20 ml de etanol de 95 por 100, v/v, recién destilado y neutralizado; valorar con solución alcohólica de hidróxido potásico 0,1 N en presencia de fenolftaleína; si el volumen utilizado de solución alcalina de superior a 0,2 ml, repetir todo el procedimiento.
22(b).5 Cálculo.
Calcular el insaponificable expresado en porcentaje.

P' = peso en g del residuo.
P = peso en g de la muestra.
22(b).6 Observaciones.
En los certificados de análisis indicar: Método del éter de petróleo.
22(b).7 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. D.5.
2. Instituto de Racionalización del Trabajo. Una Norma Española 30.305.
23. ÍNDICE DE ESCUALENO EN ACEITE DE OLIVA
23.1 Principio.
Se entiende por índice de escualeno el número de mg de este hidrocarburo contenido en 100 g de aceite.
Este método determina el índice de escualeno del aceite de oliva, o el de sus mezclas con otros aceites vegetales de composición análoga.
Los resultados obtenidos con otros aceites vegetales, con un contenido en insaponificable muy diferente al del aceite de oliva, se admitirán con reserva, no dando por segura la cifra obtenida sin asegurarse de que la separación cromatográfica de la fracción de hidrocarburos se ha verificado correctamente, comprobándose en todo caso la ausencia de esterinas, que garantiza en cierto modo la buena marcha de la operación.
23.2 Material y aparatos.
23.2.1 Matraz de saponificación, de fondo plano, de 100 ml, con refrigerante de reflujo, constituido por un tubo de 1 m de longitud.
23.2.2 Ampolla de extracción, de 500 ml de capacidad.
23.2.3 Aparato de destilación, con ajustes esmerilados y con matraz de 100 a 150 ml de capacidad.
23.2.4 Kitasato, de 250 ml.
23.2.5 Tubo de vidrio para la columna de absorción cromatográfica, con un diámetro interior de 13 mm y longitud aproximada de 400 mm. Deberá llevar en su extremo inferior un estrangulamiento o dispositivo para retener el algodón o lana de vidrio que soporta la alúmina; la parte superior irá provista de un tapón de corcho o vidrio con ajuste esmerilado, atravesado por un codo destinado a adaptar una pera de goma.
23.2.6 Embudo de vidrio de 4 cm de diámetro, aproximadamente.
23.2.7 Matraces erlenmeyer, con tapón esmerilado, de 150 ml, provistos de un codillo para destilación.
23.2.8 Buretas. Una de 25 ml, contrastada, graduada en décimas, y otra de 50 ml, graduada en décimas.
23.3 Reactivos.
23.3.1 Hidróxido potásico; reactivo para análisis.
23.3.2 Etanol de 96° exento de aldehídos
23.3.3 Éter de petróleo para análisis, p. e. 35° - 50 °C.
23.3.4 Disolución de fenolftaleína al 1 por 100 en etanol de 96°.
23.3.5 Disolución alcohólica de hidróxido potásico 0,5 N.
23.3.6 Sulfato sódico, anhidro.
23.3.7 Óxido de aluminio, para análisis cromatográficos.
23.3.8 Benceno, reactivo para análisis.
23.3.9 Cloroformo, reactivo para análisis.
23.3.10 Reactivo de Hanus. Se prepara como en 16(b).3.5.
23.3.11 Disolución de ioduro potásico al 10 por 100, exento de iodato.
23.3.12 Indicador engrudo de almidón.
23.3.13 Disolución de tiosulfato sódico 0,1 N.
23.4 Procedimiento.
23.4.1 Primera saponificación. Pesar 10 g de aceite en un matraz de 100 ml. Adicionar una disolución preparada disolviendo 3 g de hidróxido potásico en 40 ml de etanol de 96°. En general debe tomarse una cantidad de aceite que contenga de 30 a 40 mg de escualeno. Colocar el refrigerante y hervir durante una hora.
Verter la disolución de jabón, todavía caliente, en una ampolla de extracción de 500 ml; enjuagar el matraz con 100 ml de éter de petróleo e incorporarlos a la ampolla. Enfriar la ampolla y agitar fuertemente. Debe formarse una disolución homogénea. Agregar 40 ml de agua. Dejar reposar hasta la neta separación de las dos capas, aproximadamente dos horas.
Separar la capa alcohólica inferior. Pasar la capa etérea al matraz de destilación y destilar el éter.
23.4.2 Segunda saponificación. El residuo insaponificable obtenido hervirlo con 10 ml de potasa alcohólica 0,5 N, durante 20 a 30 minutos, bajo refrigerante de reflujo. Pasar la disolución alcohólica a la ampolla de extracción; agregar 50 ml de éter de petróleo y 10 ml de agua. Dejar reposar hasta separación de las dos capas; suele bastar con dos horas, aunque es más práctico dejar estar durante una noche. Separar la capa etérea y lavarla con alcohol de 50 por 100, adicionado de unas gotas de fenolftaleína, hasta eliminación total de álcali. Secar con sulfato sódico anhidro en la misma ampolla de extracción; suele bastar con 0,5 a 1 g.
Pasar la disolución de éter de petróleo por un filtro seco, recogiendo el filtrado en un matracito, tarado de 100 a 150 ml. Lavar la ampolla de extracción con 5 ml de éter de petróleo, que se hacen pasar por el mismo filtro. Lavar éste con dos porciones más, de 5 ml cada una, de éter de petróleo. Destilar el éter. Secar el residuo a 100 °C, y una vez seco, pesar Este residuo insaponificable contiene la totalidad de los hidrocarburos existentes, y una parte de las esterinas, además de otros compuestos solubles en el éter de petróleo.
23.4.3 Separación cromatográfica. Cargar el tubo de vidrio destinado a la columna de absorción con óxido de aluminio hasta una altura de 10 cm. El llenado puede hacerse en seco o en húmedo. Una vez cargada colocar un disco de papel de filtro encima de la alúmina, montándose el conjunto según se indica en 23.3.5 y 23.3.6. Agregar benceno por la parte superior de la columna, forzándolo a pasar con una ligera presión realizada con la pera de goma, hasta empapar la columna, bastando para ello unos 20 a 25 ml.
Absorbido el benceno por la alúmina, y restando sobre ésta una pequeña capa de disolvente, agregar el insaponificable disuelto en 10 ml de benceno. Lavar el matraz con 5 ml de benceno, e incorporarlo a la columna. Antes de que hayan desaparecido las últimas porciones de benceno, se agregan 55 ml de éste, y continúa el desarrollo, manteniendo la presión necesaria para que el eluyente fluya de la columna en la proporción de 2 ml por minuto aproximadamente.
Observar líquido recogido y columna bajo la luz de Wood. La disolución no deberá presentar fluorescencia marcada, como tampoco el líquido contenido en la parte inferior de la columna en una altura de 2 a 4 cm.
Pasar el líquido eluido a un matraz de 150 ml, previamente tarado. Destilar el benceno. Secar el residuo a 100 °C y pesar.
23.4.4 Valoración del escualeno. Disolver el residuo anterior en el mismo matraz con 5 ml de cloroformo; y adicionar 0,3 ml de reactivo Hanus por cada miligramo de sustancia pesada. Cerrar el matraz con el tapón de vidrio. Dejar en reposo en la oscuridad durante 15 minutos.
Añadir 5 ml de disolución de ioduro potásico al 10 por 100, y 50 ml de agua. Valorar el exceso de iodo con tiosulfato sódico 0,1 N, con engrudo de almidón como indicador.
23.4.5 Ensayo en blanco. Realizarlo en las mismas condiciones.
23.5 Cálculo.

N = normalidad del tiosulfato sódico.
V' = volumen en ml de tiosulfato sódico 0,1 N utilizados en el ensayo muestra.
V = volumen en ml de tiosulfato sódico 0,1 N utilizados en el ensayo blanco.
P = peso en g de la muestra de aceite.
23.6 Referencias.
1. J. Fitelson. J. Ass. Off. Agr. Chem. 1943. 26. 499.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.038.
24(a). DETERMINACIÓN DE LA FRACCIÓN DE ESTEROLES POR CROMATOGRAFÍA EN FASE GASEOSA
(Derogado).
24(b). DETERMINACIÓN CUANTITATIVA DE ESTEROLES
24(b).1 Principio.
Los esteroles se determinan gravimétricamente, precipitándolos como digitónidos insolubles a partir de una solución alcohólica de la grasa saponificada, para liberar los esteroles combinados.
24(b).2 Material y aparatos.
24(b).2 (1, 2 y 3) Como 24(a).2 (1, 2 y 3).
24(b).2.4 Crisol de placa filtrante, G4.
24(b).3 Reactivos.
24(b).3.1 Disolución acuosa de hidróxido potásico, aproximadamente, al 40 por 100, p/v.
24(b).3.2 Disolución digitonina al 1 por 100, p/v. en etanol de 95 por 100 v/v.
24(b).3.3 Etanol de 95 por 100, v/v.
24(b).4 Procedimiento.
Si «s» es el tanto por ciento, p/p, previsible de esteroles en la muestra, pesar exactamente, con aproximación de 0,1 por 100, 5/s g, aproximadamente, de grasa.
Agregar 3/s ml de disolución acuosa de hidróxido potásico, y después 7/s ml de etanol de 95 por 100 v/v. Adaptar el refrigerante de reflujo, y calentar a ebullición suave, sobre baño de agua hirviente, agitando de vez en cuando, hasta saponificación completa (líquido homogéneo). Continuar calentando de 15 a 20 minutos. Agregar 60 ml de agua destilada y, aproximadamente, 180 ml de etanol de 95 por 100 v/v. Calentar y mantener a, aproximadamente, 50 °C.
Agregar de 25 a 30 ml de disolución etanólica de digitonina. Dejar enfriar y reposar durante algunas horas (preferiblemente durante una noche). Filtrar la masa cristalina a través de un crisol de placa filtrante G4 con vacío, y previamente tarado con una precisión de 1 mg. Lavar con agua destilada, después con etanol de 95 por 100 v/v, y por último con algunos ml de éter etílico. Secar en estufa a 103 °C (± 2 °C) y pesar a peso constante.
24(b).5 Cálculo.

P' = peso en g de la muestra.
P = peso en g del precipitado de digitónidos.
24(b).6 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. D. 6.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.055.
25. IMPUREZAS
25.1 Principio.
Se entiende por impurezas de una grasa el conjunto de sustancias insolubles en un disolvente volátil en las condiciones descritas, y que no hayan sido ya determinadas como «agua y materias volátiles».
Del peso total obtenido de impurezas habrá que deducir, eventualmente, el peso de algunas de ellas, que, según el convenio entre las partes o según la costumbre, no deban ser consideradas como tales.
Los jabones de cal se considerarán como impurezas, a menos que por acuerdo entre las partes se decida otra cosa. En este último caso no será considerado como impureza más que la cal que contenga, expresada en óxido de calcio.
Salvo indicaciones contrarias, los jabones alcalinos no serán considerados como impurezas más que por las bases que contengan, expresadas en óxidos.
25.2 Reactivos.
Para la determinación se podrá usar cualquiera de los disolventes citados posteriormente, condicionando su empleo a la naturaleza de la materia grasa tratada, y a la de las sustancias que deban ser consideradas como impurezas en relación con el destino o aplicación que deba darse a la grasa. Dicha elección podrá ser impuesta por razones o convenio entre las partes.
Las impurezas de las materias grasas comestibles y de las materias grasas brutas con destino comestible después de tratamiento adecuado se determinarán con éter de petróleo.
25.2.1 Éter de petróleo puro para análisis. En las condiciones del procedimiento descrito, el éter de petróleo deja insolubles: Impurezas mecánicas (tierra, arena y residuos diversos); parte de ácidos oxidados libres y sus productos de polimerización, lactonas-jabones de cal, hidratos de carbono, materias nitrogenadas, determinadas resinas, materias minerales, y no disuelve más que parcialmente los jabones alcalinos.
25.2.2 Éter técnico (C2H5)2O, puro para análisis. En las condiciones del procedimiento descrito, el éter etílico deja insolubles: Las impurezas mecánicas (tierra, arena y residuos diversos), materias minerales, hidratos de carbono, materias nitrogenadas, ciertas resinas, jabones de cal y jabones alcalinos.
25.2.3 Sulfuro de carbono, S2C, puro para análisis recién destilado. En las condiciones del procedimiento descrito, el sulfuro de carbono deja insolubles: Impurezas mecánicas (tierra, arena y residuos diversos), materias minerales, hidratos de carbono, materias nitrogenadas, ciertas resinas y jabones de cal. No disuelve más que parcialmente los jabones alcalinos.
25.3 Procedimiento.
Pesar exactamente, con una aproximación de 0,01 g, aproximadamente 20 g de materia grasa en un erlenmeyer, con tapón de vidrio esmerilado (la grasa solamente fundida, si es necesario) agregar 200 ml del disolvente elegido, tapar, agitar y dejar en reposo a, aproximadamente, 20 °C durante 30 minutos, si se trata de éter etílico o éter de petróleo, y 12 horas si se trata de sulfuro de carbono.
Filtrar sobre filtro sin cenizas, de 12 cm de diámetro, previamente secado y tarado, lavar el filtro con pequeñas porciones de disolvente, utilizando la cantidad estrictamente necesaria para que el filtrado esté exento de materia grasa, retirar el filtro del embudo, dejar evaporar el disolvente al aire libre, y terminar la evaporación en estufa a 103 °C (± 2 °C). Pesar el filtro.
25.4 Cálculo.

P" = peso en g del filtro seco.
P'= peso en g del filtro más impurezas.
P = peso en g de la muestra.
Indicar el disolvente utilizado y, eventualmente, los componentes que han sido deducidos de las impurezas.
25.5 Observaciones.
25.5.1 Si el filtrado contiene cenizas, eliminar el disolvente por destilación, incinerar el residuo, pesar y agregar esta pesada a las impurezas del filtro.
25.5.2 Para una determinación de gran exactitud deberá emplearse una cantidad de disolvente cincuenta veces superior al peso de grasa tomada.
25.5.3 En el caso de una materia grasa que contenga jabones, y en el caso de que estos jabones no deban ser considerados como impurezas, más que por las bases que contengan, separar el residuo insoluble del filtro y tratarlo a ebullición con refrigerante de reflujo, con ácido clorhídrico diluido (1:5), hasta que los jabones se hayan descompuesto totalmente. Extraer la materia grasa así liberada en una ampolla de decantación por medio del disolvente utilizado para la determinación de las impurezas. Después de pequeños lavados con agua, destinados a eliminar la acidez mineral, filtración y eliminación del disolvente, pesar los ácidos grasos, y deducir de las impurezas el tanto por ciento así obtenido.
25.6 Referencias.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. C. 2.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.002.
26. CENIZAS
26.1 Principio.
Las cenizas representan el residuo mineral de las materias grasas, previamente filtradas.
Su tanto por ciento no debe añadirse al de las impurezas insolubles en un disolvente, para evitar que ciertos elementos sean considerados dos veces. Ahora bien, como continuación a la dosificación de las impurezas, se puede llegar a dosificar las cenizas sobre la materia grasa purificada después de la expulsión del disolvente. En tal caso, el tanto por ciento de cenizas se añade al de impurezas.
26.2 Material y aparatos.
26.2.1 Cápsula de incineración de, aproximadamente, 50 ml de capacidad.
26.3 Procedimiento.
Pesar exactamente, con una aproximación de 0,01 g, aproximadamente 10 g de materia grasa en la cápsula, previamente tarada.
Calentar prudentemente hasta el punto de inflamación y dejar arder a la sustancia espontáneamente, calcinar moderadamente hasta la obtención de un residuo que no emita más vapores, tomar el residuo con agua destilada, filtrar con la ayuda eventual de una ligera corriente de oxígeno o con algunas gotas de agua oxigenada, dejar enfriar la cápsula, verter cuantitativamente el filtrado anterior, evaporar a sequedad en baño de agua. Después calcinar, aproximadamente, a 400 °C y pesar.
Si fuera necesario se pueden recarbonatar las cenizas con carbonato amónico, o agua saturada de ácido carbónico.
26.4 Cálculo.

P' = peso en g de las cenizas.
P = peso en g de la muestra ensayada.
26.5 Referencia.
1. International Union of Pure and Applied Chemistry. Standard Methods for the Analysis of Oils, Fats and Soaps, 1964, II. C. 3.
27. DETECCIÓN DE COMPUESTOS CLORADOS
27.1 Principio.
Este método determina cualitativamente la presencia de cloro procedente de solventes clorados u otros compuestos.
Es aplicable a los aceites vegetales normales.
27.2 Material y aparatos.
27.2.1 Alambre de cobre puro, de aproximadamente 1 mm de diámetro; bien pulido y acoplado a un soporte aislante del calor.
27.2.2 Mechero Bunsen, o quemador de gas equivalente
27.3 Procedimiento.
Calentar el alambre de cobre en la llama del Bunsen hasta que no sea visible color verde.
Mojar la parte calentada del alambre con la muestra de aceite y volver a introducir dicha parte en la llama; si ésta presenta un color verde definido, indica la presencia de cloro en la muestra.
27.4 Expresión de resultados.
Coloración verde, más o menos intensa: Positivo. Coloración no verde: Negativo.
27.5 Referencia.
1. American Oil Chemists' Society. Official and Tentative Methods. Ca. 7.35.
28(a). RECONOCIMIENTO DE AZUFRE
(Ensayo del benzoato de plata)
28(a).1 Principio.
Este método determina cualitativamente la presencia de azufre.
Aplicable a la detección de azufre del sulfuro de carbono en la extracción del aceite de orujo de oliva.
28(a).2 Material y aparatos.
28(a).2.1 Tubos de ensayo de tamaño conveniente.
28(a).2.2 Baño de aceite, capaz de calentar la muestra a 150 °C en cinco minutos.
28(a).3 Reactivos.
28(a).3.1 Benzoato de plata: Mezclar cantidades equivalentes de nitrato de plata y benzoato sódico, filtrar, lavar el precipitado con varias porciones de agua destilada, secar en estufa a 60-70 °C. Preservarlo de la luz. Pulverizar el benzoato de plata seco, antes del uso,
28(a).4 Procedimiento.
Filtrar la muestra, tomar en dos tubos de ensayo 5 ml en cada uno, calentar en el baño de aceite, alcanzando 150 °C en, aproximadamente, cinco minutos, para evitar sobrecalentamientos
Retirar del baño uno de los tubos, agregarle aproximadamente 0,2 g de benzoato de plata y agitar inmediatamente. Observar el aspecto del tubo, comparándolo con el de ensayo en blanco. La aparición de un ennegrecimiento indica la presencia de azufre.
28(a).5 Referencia.
1. American of Chemists' Society, Official and Tentative Methods. Ca. 8b. 35.
28(b). RECONOCIMIENTO DE AZUFRE
(Ensayo de la moneda)
28(b).1 Principio.
Este método determina cualitativamente la presencia de azufre.
Aplicable a la detección de azufre en el sulfuro de carbono empleado en la extracción del aceite de oliva.
28(b).2 Material y aparatos.
28(b).2.1 Tubo de ensayo o vaso de tamaño conveniente.
28(b).2.2 Varilla o bolas de vidrio para soportar la moneda.
28(b).2.3 Una moneda pulida, preferiblemente nueva.
28(b).2.4 Baño de aceite capaz de calentar la muestra a 210 °C en unos 30 minutos.
28(b).3 Procedimiento.
Colocar la moneda en un tubo de ensayo o vaso, soportada por bolas o trozos de varilla de vidrio. Agregar 20 ml de la muestra, que han de cubrir la moneda.
Colocar el tubo de ensayo o vaso en el baño de aceite, calentar de forma que la muestra alcance 210 °C en, aproximadamente, 30 minutos, mantener a esta temperatura durante 10 minutos más.
Enfriar y retirar la moneda, colocar sobre una superficie limpia y examinar cuidadosamente si presenta despulido o ennegrecimiento.
Si éste es evidente, la presencia de azufre es positiva.
28(b).4 Referencia.
1 American Oil Chemists' Society. Official and Tentative Methods. Ca. 8a. 35.
29. DETERMINACIÓN DE JABÓN EN ACEITE DE OLIVA
29.1 Principio.
Este método tiene por objeto el reconocimiento de jabones alcalinos, magnésicos o cálcicos, e identificación de los iones de estos últimos elementos en aceites de oliva que hayan sido total o parcialmente neutralizados con compuestos básicos de estos metales y sometidos a una decantación o centrifugación.
29.2 Material y aparatos.
29.2.1 Ampolla de decantación de 100 ml, con tapón esmerilado.
29.2.2 Probeta graduada, de 10 ml.
29.2.3 Embudo de filtración de 5 a 6 cm de diámetro.
29.2.4 Cápsulas de vidrio Pyrex o Jena, redondas o fondo plano, de 5 a 6 cm de diámetro.
29.2.5 Tubos de ensayo, de 100 mm de longitud y 10 mm de diámetro.
29.2.6 Cápsulas redondas de porcelana de 4 a 5 cm de diámetro.
29.2.7 Vidrios de reloj de 4 cm de diámetro.
29.2.8 Papel de filtro sin cenizas de 9 cm de diámetro.
29.3 Reactivos.
29.3.1 Ácido clorhídrico, reactivo para análisis, al 10 por 100.
29.3.2 Ácido nítrico, reactivo para análisis, 2 N.
29.3.3 Hidróxido sódico, reactivo para análisis, 2 N.
29.3.4 Nitrato de plata, reactivo para análisis, al 5 por 100.
29.3.5 Quinalizarina, reactivo para análisis, disolución saturada en etanol de 96°, recientemente preparada.
29.3.6 Sal sódica de la osazona del ácido dihidroxitartárico.
29.4 Procedimiento.
29.4.1 Extracción. Medir 10 ml de aceite con la probeta graduada y verterlos en la ampolla de decantación, previamente mojada con agua. Añadir 10 ml de disolución de ácido clorhídrico. Agitar durante 15 ó 20 segundos. Dejar reposar hasta separación de las dos capas, aproximadamente de 10 a 15 minutos.
29.4.2 Centrifugación. La capa acuosa decantada centrifugada entre 4.000 y 5.000 r.p.m. Si no se dispone de centrífuga, filtrar a través de papel de filtro, sin cenizas, previamente humedecido.
29.4.3 Ensayo general de jabones. Verter en cápsulas de vidrio 2 ml de la disolución filtrada o centrifugada. Evaporar a sequedad en bario de arena o de agua. Disolver el residuo con unas gotas de agua destilada y volver a evaporar. Repetir esta operación dos veces. Agregar el residuo de la tercera evaporación, una gota de ácido nítrico y 2 ml de agua destilada, calentar ligeramente hasta disolver el residuo, pasar a un tubo de ensayo, agregar tres gotas de disolución de nitrato de plata. Un precipitado coposo de cloruro de plata o una turbidez para pequeñas cantidades, revela la existencia de jabones en el aceite.
Realizar el ensayo en blanco, evaporando 2 ml de la disolución de ácido clorhídrico utilizada, o mejor todavía operando sobre un aceite virgen garantizado. El resultado se interpretará comparando la turbidez obtenida en el ensayo en blanco con la del problema. Ligeras diferencias no deben interpretarse positivamente.
29.4.4 Identificación del magnesio. Verter 15 gotas de la disolución filtrada en 29.4.2 en una cápsula de porcelana de fondo redondo, agregar dos gotas de disolución de quinalizarina, agitar mediante ligero movimiento de rotación, agregar, gota a gota, disolución de hidróxido sódico, agitando a cada adición con movimiento de rotación, hasta que se produzca un cambio de color y después tres o cuatro más. La existencia de magnesio origina un precipitado fino de color azul; la ausencia de magnesio se acusa por no producirse el color azul y quedar la solución de un color violeta azulado.
Paralelamente, realizar un ensayo en blanco con la disolución de ácido clorhídrico. Un resultado positivo obliga a preparar otra disolución de ácido clorhídrico y repetir la extracción de la muestra como en 29.4.1.
29.4.5 Identificación del calcio. Depositar 0,5 ml de la disolución filtrada 29.4.2 en una cápsula de vidrio, neutralizar lo más exactamente posible con la disolución de hidróxido sódico, utilizando papel de tornasol. Tomar dos gotas en un vidrio de reloj, agregar dos o tres granitos del reactivo sólido 29.3.6. El reactivo se difundirá a través de la gota, comunicando a ésta un color naranja amarillento. Si al cabo de diez o quince minutos la disolución permanece transparente, no existe calcio. Si la disolución se enturbia, formándose un precipitado amarillo, se acusa la presencia de calcio.
Realizar el ensayo en blanco con la disolución clorhídrica, repitiendo las operaciones de 29.4.5. Un resultado positivo obliga a preparar otra disolución de ácido clorhídrico y repetir las operaciones indicadas en 29.4.1.
29.5 Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.039.
30. RECONOCIMIENTO DE JABÓN EN ACEITE REFINADO
30.1 Principio.
Este método tiene por objeto identificar la presencia de jabón en aceites refinados.
30.2 Material y aparatos.
30.2.1 Tubos de ensayo de 150 x 15 mm.
30.3 Reactivos.
30.3.1 Disolución al 0,1 por 100 de azul de bromofenol en etanol de 96°.
30.3.2 Acetona, recientemente destilada, con un 2 por 100 de agua.
30.4 Procedimiento.
Los tubos de ensayo deben limpiarse escrupulosamente antes del uso. La acetona al 2 por 100 de agua con la adición de algunas gotas de la disolución de azul de bromofenol debe dar coloración del amarillo al amarillo-verdoso; si adquiere un matiz verdoso, repetir la operación.
Colocar en un tubo de ensayo 10 ml de acetona y una gota de disolución de azul de bromofenol; la disolución debe quedar amarilla. Agregar 10 g de aceite, cerrar con un tapón limpio, agitar y dejar decantar. La capa acetónica superior no debe presentar una coloración azul, ésta revelaría la presencia de jabón.
30.5 Expresión de los resultados.
30.5.1 Coloración amarilla: negativa. Coloración azul: positiva.
30.5.2 Observando por reflexión, color azul ultramar claro, y por transparencia, color azul violeta intenso; contenido de jabón superior al 0,001 por 100, expresado en hidróxido sódico.
30.5.3 Capa acetónica con coloración verde claro: contenido de jabón comprendido en 0,001 por 100 y 0,0001 por 100.
30.6 Observaciones.
Para que el ensayo tenga la sensibilidad indicada, el aceite debe tener una acidez libre inferior al 1 por 100 (expresada en ácido oleico).
30.7 Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.029.
31. ABSORCIÓN ESPECTROFOTOMÉTRICA ULTRAVIOLETA
31.1 Principio.
Este método se fundamenta en la medida espectrofotométrica ultravioleta del coeficiente de extinción a las longitudes de onda 232 y 270 mμ representados por K23 2 y K2 7 0, respectivamente.
Ambos coeficientes se emplean como criterio de calidad de los aceites.
31.2 Material y aparatos.
31.2.1 Espectrofotómetro ultravioleta. La escala de longitudes de onda permitirá medir entre 220 y 350 nm con precisión de 1 nm.
31.2.2 Cubeta prismática de cuarzo, de 1 cm de espesor o paso ópticamente transparente entre 220 y 350 nm. La transmisión a 220 nm será, como mínimo, de un 80 por 100. Las cubetas en uso tendrán, entre sí, una desviación no superior al 2 por 100 a 220 nm.
31.2.3 Matraces aforados con tapón de vidrio esmerilado de 10, 500 y 1.000 ml.
31.3 Reactivos.
31.3.1 Ciclohexano. Para espectrofotometría, cuyos valores minutos de transmisión serán: 40 por 100 a 220 nm y 95 por 100 a 250 nm.
31.3.2 Disolución de hidróxido potásico, para análisis, 0,05 N.
31.3.3 Disolución patrón de cromato potásico. Disolver 2.000 miligramos de cromato potásico, para análisis, en 1 litro de disolución de hidróxido potásico 0,05 N. Tomar 25 ml de esta solución en un matraz aforado de 500 ml, completando hasta el enrase con disolución de hidróxido potásico 0,05 N.
La densidad óptica de esta última disolución a 275 nm, medida en cubeta de 1 cm, tomando como referencia la disolución de hidróxido potásico 0,05 N, deberá ser de 0,200 ± 0,005.
31.4 Procedimiento.
Comprobar la escala de transmisión a absorbancia.
Si el aceite no está completamente limpio y transparente se filtra a la temperatura ambiente, a través de papel de filtro adecuado.
Pesar exactamente, en un matraz aforado de 10 ml, aproximadamente, las siguientes cantidades de muestra: 90 mg de aceite de oliva virgen, 60 mg de aceite refinado o su mezcla con vírgenes, 40 mg de aceite de orujo bruto o refinado, 40 miligramos de aceite de semilla; disolver en ciclohexano y completar hasta el enrase
En cubeta de 1 cm de paso, medir la extinción a 232 y 270 nm utilizando ciclohexano como patrón de comparación.
La lectura debe estar comprendida entre 0,2 y 0,8 de absorbancia; en caso contrario se repetirá la medida, bien diluyendo convenientemente o procediendo a una nueva pesada.
31.5 Cálculo.

 λ = extinción específica a la longitud de onda λ.
λ = extinción específica a la longitud de onda λ.
A λ = absorción leída en el espectrofotómetro.
c = concentración de la disolución en g/100 ml.
e = espesor en cm de la cubeta.
31.6 Observación.
Los aceites de oliva vírgenes que sea necesario purificar para realizar esta determinación se someterán al siguiente tratamiento en columna de alúmina:
Preparar una columna rellenando con 30 g de alúmina activada, para cromatografía, de actividad I, según Brockman, un tubo de vidrio para cromatografía, cuya parte superior tenga 230 mm de longitud y 35 mm de diámetro interior, y la parte inferior sea de la misma longitud y 10 mm de diámetro interior. Esta parte inferior estará provista de un estrangulamiento o dispositivo para retener el algodón o lana de vidrio que actúa como retén o soporte del absorbente. La alúmina activada ocupará aproximadamente toda la parte inferior de la columna, y puede adicionarse seca o humedecida con el disolvente, que puede ser éter de petróleo ligero o ciclohexano para cromatografía.
Verter en la columna una disolución de 10 g de aceite, pesados con una aproximación de 0,01 g, en 100 ml de disolvente. Recoger la solución oleosa a la salida de la columna sobre un filtro Jena de placa filtrante, previamente secado en estufa a 105 °C, recogiendo el filtrado en un matraz pequeño. Destilar el disolvente en vacío, a temperatura inferior a 25 °C.
El aceite libre de disolvente se utiliza inmediatamente para determinar los coeficientes de extinción en el ultravioleta.
31.7 Referencias.
1 J. P. Wolf. Rev. Francaise Corps Gras, 1954. 1, 215.
2 Consejo Oleícola Internacional, 1957.
32(a). ÍNDICE DE BELLIER
32(a).1 Principio.
El índice de Bellier de un aceite es la temperatura a la que se produce la precipitación de las sales de los ácidos grasos de dicho aceite cuando está saponificado y disuelto con arreglo a las condiciones de este método.
32(a).2 Material y aparatos.
32(a).2.1 Tubos de ensayo de 26-27 mm de diámetro y 220 mm de longitud.
32(a).2.2 Refrigerante constituido por un tubo de vidrio de aproximadamente 800 mm de longitud con un tapón en el extremo.
32(a).2.3 Termómetro graduado de 0 °C a 50 °C, con divisiones de décimas de grado, adaptado a un tapón que sirve de cierre al tubo de ensayo.
32(a).2.4 Pipetas graduadas.
32(a).3 Reactivos.
32(a).3.1 Disolución hidro-alcohólica de hidróxido potásico. Disolver 42,5 g de KOH puro en 72 ml de agua destilada y llevar a 500 ml con etanol de 95°.
32(a).3.2 Disolución de alcohol etílico: título 70° (utilizar alcohol etílico puro indiferente a los hidróxidos alcalinos).
32(a).3.3 Disolución acuosa de ácido acético: 1 + 2 (en volúmenes); ajustada de manera tal que 1,5 ml neutralicen exactamente (indicador fenolftaleína), 5 ml de la disolución hidroalcohólica de hidróxido potásico.
32(a).4 Procedimiento.
Eliminar el agua del aceite por decantación y filtración sobre papel, efectuada a una temperatura ligeramente superior al punto de fusión de determinados componentes sólidos que hubieran podido separarse de la materia grasa fluida.
Colocar en un tubo de ensayo 1 ml de grasa y 5 ml de disolución de potasa. Cubrir con el refrigerante y calentar moderadamente agitándolo por rotación, de cuando en cuando, hasta completar saponificación, es decir, hasta obtener una disolución perfectamente límpida.
Dejar enfriar. Separar el refrigerante. Añadir 1,5 ml de la disolución de ácido acético y 50 ml de la disolución de alcohol de 70°. Colocar el termómetro y homogeneizar.
Poner el tubo en un vaso lleno de agua a una temperatura de 23-25 °C. En el caso de que se produzca un precipitado en forma de copos, dejar durante una hora a dicha temperatura y filtrar en un tubo de ensayo.
Introducir el termómetro en el tubo de ensayo que tiene la disolución transparente filtrada. Colocar unos instantes en un vaso de agua cuya temperatura sea inferior a 10 °C, aproximadamente, a la supuesta para el índice de Bellier.
Retirarlo y homogeneizar la temperatura, para lo cual se agita dicho tubo invirtiéndolo (la velocidad de enfriamiento debe ser aproximadamente 1 °C por minuto); repetir esta operación tantas veces como sea necesario hasta que aparezca turbidez. En ese momento anotar la temperatura.
Dejar que la temperatura de la disolución aumente algunos grados con el fin de disolver el precipitado. Homogeneizar invirtiendo el tubo y enfriar. El enfriamiento debe ser lento y las agitaciones más frecuentes a medida que la temperatura se aproxima a la apuntada en el ensayo anterior. La temperatura que servirá para fijar el índice de Bellier será la correspondiente a la reaparición de la turbidez.
32(a).5 Cálculo.
Índice de Bellier (ácido acético) = t °C
32(a).6 Observaciones.
Los resultados de dos determinaciones paralelas no deben variar en más de 0,25 °C.
32(a).7 Referencias.
1. Bellier.‒Copt. Rend. Congr., Intern. Chim. Appl. 1896. 4, 311.
2. Marcille, R.‒Ann. Chim. Anal. Chim. Appl. 1939. 21, 311.
3. Consejo Oleícola Internacional, 1957.
32(b). ÍNDICE DE BELLIER
(Método ácido clorhídrico)
32(b).1 Principio.
Como 32(a).1.
32(b).2 Material y aparatos.
Como 32(a).2.
32(b).3 Reactivos.
32(b).3.1 Solución hidro-alcohólica de hidróxido potásico. Disolver 10 g de KOH en 100 ml de etanol.
32(b).3.2 Disolución de alcohol etílico; título 70° (utilizar etanol puro indiferente a los hidróxidos alcalinos).
32(b).3.3 Solución de ácido clorhídrico (d = 1,16). Diluir 83 ml de ácido clorhídrico concentrado (d = 1,19) a 100 ml de agua.
32(b).4 Procedimiento.
Como en 32(a).4, excepto que se saponificará con solución alcalina 32(b).3.1, y se emplearán 0,8 ml de disolución ácida 32(b).3.3, en lugar de los 1,5 ml de disolución de ácido acético.
32(b).5 Cálculo.
Índice de Bellier (método ácido clorhídrico) = t °C
32(b).6 Referencias.
1. J. Assoc. Offic. Agr., 28, 293 (1945); 32, 363 (1949).
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.009.
33. RECONOCIMIENTO DE ACEITE DE ORUJO DE ACEITUNA
(Bellier-Marcille)
33.1 Principio.
Este método se emplea para detectar la presencia de aceite de orujo de aceituna en mezcla con aceites de oliva.
33.2 Material y aparatos.
33.2.1 Matraz de 100 ml provisto de refrigerante de reflujo, con cierres esmerilados.
33.2.2 Pipeta de 5 ml graduada en décimas de grado.
33.2.3 Pipeta de 5 ml.
33.2.4 Probeta de 50 ml.
33.2.5 Bloque de calefacción que permita mantener el matraz a unos 80 °C.
33.2.6 Termómetro de 0 °C a 60 °C, graduado en décimas.
33.3 Reactivos.
33.3 (1, 2 y 3). Como en 32.3 (1, 2 y 3).
33.4 Procedimiento.
Preparar la muestra como en 32.4.
Verter en el matraz 1 g aproximadamente de aceite. Agregar 5 ml de disolución alcohólica de hidróxido potásico. Ajustar el refrigerante y mantener a ebullición durante diez minutos, agitando de cuando en cuando, dejar enfriar hasta la temperatura ambiente, agregar 1,5 ml de disolución de ácido acético y 50 ml de etanol de 70°, calentando previamente a 50 °C. Mezclar mediante agitación, introducir el termómetro y dejar enfriar, observando el aspecto de la disolución cuando alcance la temperatura de 45 °C. Si se forma un precipitado floconoso a temperatura superior a 40r C, el ensayo es positivo.
Dejar enfriar a temperatura ambiente (no inferior a 18 °C) durante 12 horas, como mínimo. Observar de nuevo la disolución: La formación de un precipitado floconoso, flotando en el centro del líquido, indica que la reacción es positiva. Una turbidez no resuelta en copos no indica la presencia de aceite de orujo.
33.5 Expresión de los resultados.
Positivo o negativo.
33.6 Observación.
Excepcionalmente, puede suceder que algunos aceites de oliva vírgenes, de segunda presión, den un resultado positivo.
33.7 Referencias.
1. Bellier.‒Compt. Rend. Congr. Inter. Chim. Appl. 1896, 4, 311.
2. Marcille, R.‒Ann. Chim. Anal. Chim. Appl. 1939, 21, 311.
3. Consejo Oleícola Internacional, 1957.
34. DETERMINACIÓN CUALITATIVA Y CUANTITATIVA DE LOS ÁCIDOS GRASOS SITUADOS EN LA POSICIÓN B DE LOS TRIGLICÉRIDOS
34.1 Principio.
El procedimiento se basa en someter la muestra de grasa neutra, previamente purificada mediante tratamiento con alúmina activada, a una hidrólisis bajo la acción de la lipasa pancreática, que actúa selectivamente sobre los radiales acilo situados en la posición α de los triglicéridos, con una acumulación de β-monoglicéridos inalterados.
Se separan estos β-monoglicéridos por cromatografía en capa fina de gel de sílice, efectuándose el análisis cualitativo y cuantitativo de los ácidos por cromatografía en fase gaseosa de sus ésteres metílicos.
Es aplicable a los aceites y grasas comestibles que se encuentran normalmente en el comercio tanto al estado bruto como refinados.
Es utilizable, con reserva, en grasas que contengan ácidos con funciones oxigenadas o de cadena no lineales, cuyo comportamiento ante la lipasa ha de ser previamente investigado.
34.2 Material y aparatos.
34.2.1 Tubo para cromatografía, el cual consta de dos partes: La parte superior tiene un diámetro interior de 35 mm, y la parte inferior, un diámetro interior de 10 mm. Cada una de estas partes tiene una longitud de 23 cm, lo que da al tubo cromatográfico una longitud total de 46 cm. La mitad inferior tiene, en su extremidad, una estrangulación o el dispositivo conveniente para retener el tapón de algodón o lana de vidrio que ha de servir de soporte al absorbente; o bien, una placa filtrante de vidrio de porosidad 2.
34.2.2 Filtro de vidrio Jena 11 G2 u otro equivalente.
34.2.3 Matraz redondo de fondo plano y boca esmerilada, de 100 ml de capacidad.
34.2.4 Ampolla de extracción de 500 ml.
34.2.5 Vasito de vidrio, cilíndrico de fondo plano y boca esmerilada, con las siguientes dimensiones aproximadas: 18 milímetros ∅; 60 mm de altura total, incluida la boca, constituida por un cono esmerilado, normalizado, con una altura de 10 milímetros; irá provisto de tapón de teflón, aunque puede sustituirse por tapón de vidrio.
34.2.6 Pipetas de 0,2-0,5-1 y 2 ml de capacidad.
34.2.7 Vibrador en el cual pueda agitarse fuertemente el vaso descrito en 34.2.5; debe dar, aproximadamente, unas 50 vibraciones por segundo.
34.2.8 Equipo de cromatografía en capa fina; placas de gel de sílice G de 20 x 20 cm y 0,25 mm de espesor y cubeta para el desarrollo de dichas placas.
34.2.9 Matraz de 50 ml de forma análoga a un matraz aforado con cuello de 10 mm ∅ exterior y boca esmerilada, cono normalizado 12/21.
34.2.10 Embudo de vidrio soplado.
34.2.11 Varilla de reflujo, provista de cono esmerilado macho 12/21.
34.2.12 Medidor de pH de lectura directa. En el caso de operar con indicador interno según se indica en 34.5.1, puede prescindirse de este aparato, aunque es preferible su utilización.
34.2.13 Termostato de agua que permita mantener una constancia de temperatura ± 0,5 °C.
34.2.14 Bureta de 5 ml con divisiones de 1/20 ml, y bureta de 25 ml graduada en décimas.
34.2.15 Vaso de 50 ml de capacidad.
34.2.16 Agitador pequeño de hélice.
34.2.17 Cronómetro.
34.3 Reactivos.
34.3.1 Alúmina activada, para cromatografía, actividad I según Brockman. Un tipo de alúmina que da muy buenos resultados es el «óxido de aluminio FLUKA para cromatografía», tipo 507C neutro.
34.3.2 Etanol de 96° exento de aldehídos.
34.3.3 Disolución de hidróxido sódico 2N. Disolver 100 g de hidróxido sódico en lentejas, calidad R. A., en agua destilada, recientemente hervida, completando el volumen hasta 1.000 mililitros.
34.3.4 Lipasa pancreática de cerdo, cuya actividad esté comprendida entre 0,8 y 2 unidades lipásicas por mg.
34.3.5 Disolución acuosa de Tris (hidroximetil) ‒amino-metano 1 M‒ ajustada a pH = 8 con ácido clorhídrico 6N.
34.3.6 Disolución acuosa de cloruro cálcico al 22 por 100.
34.3.7 Disolución acuosa de colato sódico al 0,1 por 100.
34.3.8 Ácido clorhídrico, aproximadamente 6 N.
34.3.9 Éter etílico.
34.3.10 Éter de petróleo, o hexano normal.
34.3.11 Mezcla de éter de petróleo-éter etílico, en la proporción de 2 a 1, a la cual se adiciona 1,6 por 100 de ácido fórmico (aproximadamente de 98 por 100).
34.3.12 Disolución de metilato sódico en metanol. Disolver sodio en metanol, con las precauciones habituales en la cuantía necesaria para obtener una concentración 0,2 M.
34.3.13 Disolución de ClH gaseoso en metanol aproximadamente al 4 por 100.
34.3.14 Disolución acuosa saturada de cloruro sódico.
34.3.15 Suspensión de goma arábiga al 10 por 100 en agua.
34.3.16 Disolución acuosa de colato sódico al 20 por 100.
34.3.17 Hidróxido sódico 0,1 N.
34.3.18 Disolución acuosa de rojo cresol al 1 por 100.
34.3.19 Aceite de oliva neutralizado.
34.3.20 Fenolftaleína al 1 por 100 en metanol.
34.3.21 Acetona o cloroformo, calidad para análisis.
34.4 Procedimiento.
Preparación de la lipasa pancreática: Aunque este producto puede adquirirse en el mercado, damos a continuación un método para su preparación: 5 kg de páncreas frescos de cerdo, se enfrían a 0 °C; se liberan de la grasa sólida y del tejido conjuntivo que los rodean, y se trituran en un molino de cuchillas hasta obtener una pasta fluida. Esta pasta se agita en frío, durante 4-6 horas, con 2,5 litros de acetona anhidra y después se centrifuga. El residuo se extrae otras tres veces con el mismo volumen de acetona, dos veces con una mezcla de acetona-éter etílico (1:1) y dos veces con éter etílico. El producto se seca durante 48 horas al vacío, obteniéndose un polvo estable que debe conservarse en refrigerador.
Valoración de la actividad lipolítica del polvo de lipasa: Se prepara una emulsión de aceite de oliva agitando, en un mezclador adecuado durante unos 10 minutos, una mezcla constituida por 165 ml de suspensión de goma arábiga al 10 por 100; 15 g de hielo picado en trozos finos; y 20 ml de aceite de oliva, previamente neutralizado.
Diez ml de la emulsión anterior se introducen en un vaso de 50 ml. Se añaden 0,3 ml de disolución de colato sódico al 20 por 100 y 20 ml de agua destilada.
Se coloca el vaso en el termostato, regulado a 37 °C, y se introducen los electrodos de un medidor de pH y un agitador de hélice (34.5.1).
Con la bureta de 5 ml se añade, gota a gota, disolución de hidróxido sódico 0,1 N hasta conseguir un pH de 8,5. Se añade un volumen conveniente de suspensión de polvo de lipasa en agua, según se indica en el párrafo siguiente y, tan pronto como el medidor de pH acuse un pH de 8,3, se pone en marcha el cronómetro y se va añadiendo la disolución de hidróxido sódico 0,1 N, al ritmo necesario para mantener constante el valor 8,3 de pH, anotando cada minuto el volumen de disolución alcalina consumida que acusa la bureta.
Los datos obtenidos se llevan a un sistema de ejes coordenados, marcando, en abscisas, los tiempos y, en ordenadas, los mililitros de disolución alcalina consumida para mantener el pH constante; debe obtenerse una gráfica lineal.
La suspensión de lipasa a que se alude en el párrafo anterior, se prepara al 0,1 por 100 en agua, tomando para cada ensayo la cantidad necesaria para que en 4 ó 5 minutos se consuma alrededor de 1 ml de disolución alcalina. Normalmente se consigue este resultado con 1 a 5 mg de polvo.
La unidad lipásica se define como la cantidad de enzima que libera 10/μ-equiv, de ácido por minuto. La actividad del polvo utilizado, medida en unidades lipásicas por mg, se calcula por la fórmula:

A = actividad en unidades lipásicas por mg
V = ml de disolución de hidróxido sódico 0,1 N, consumidos por minuto, calculados a partir de la gráfica.
P = peso tomado de polvo, expresado en mg.
La lipasa utilizada ha de tener una actividad comprendida entre 0,8 y 2 unidades lipásicas por miligramo.
Preparación de la muestra. Si la grasa tuviera una acidez libre igual o inferior a 3 por 100, se someterá directamente a la purificación en columna de alúmina, según se describe más adelante.
Si la grasa tuviese una acidez superior al 3 por 100 se someterá a una neutralización alcalina en hexano, según se describe más adelante, y la grasa neutra obtenida se purificará en columna de alúmina siguiendo el procedimiento descrito a continuación:
Purificación con alúmina: Se toman 15 g de alúmina activada y se disponen en el tubo cromatográfico (34.2.1), alojándose en la mitad inferior de diámetro más estrecho, pudiéndose efectuar el llenado en seco o en húmedo, utilizando para ello el disolvente que se disponga para el desarrollo, bien sea el éter de petróleo o el hexano. Una vez preparada la columna, se vierten 5 g de muestra, pesada con exactitud del centigramo y disuelta en 50 ml de éter de petróleo o hexano (34.5.2).
La disolución oleosa que pasa por la columna se filtra a través del filtro de vidrio (34.2.2) previamente desecado en estufa a 105 °C.
La disolución filtrada se recoge en un matracito redondo de unos 100 ml y se destila el disolvente al vacío, operando a una temperatura no superior a 30 °C. Para esta operación resulta muy ventajosa la utilización de un evaporador rotatorio (34.5.3).
El aceite obtenido, libre de disolvente, se utilizará para la hidrólisis por la lipasa pancreática, según se describe más adelante.
Neutralización con hidróxido sódico en hexano. En una ampolla de extracción de 500 ml se introducen unos 20 g de muestra, 100 ml de hexano, 50 ml de etanol, una gota de disolución alcohólica de fenolftaleína al 1 por 100, y el volumen de disolución de hidróxido sódico 2N, correspondiente a la acidez libre de la muestra, medido con pipeta o bureta graduada en décimas de milímetro (34.5.4).
Se agita la mezcla enérgicamente; se agregan 50 ml de agua destilada, se agita nuevamente y se deja en reposo hasta que la mezcla se separe en dos capas bien definidas.
Se extrae por la llave la capa hidroalcohólica inferior conteniendo los jabones.
La disolución de hexano conteniendo la grasa neutra se lava con porciones sucesivas de 25 ml de una mezcla de etanol de 96° y agua destilada 1:1 (v/v), hasta que el líquido de lavado no coloree en rosa a la fenolftaleína.
La disolución en hexano, libre de jabones, se deseca agregando 3-4 gramos de sulfato sódico anhidro; y se evapora el disolvente en evaporador rotatorio al vacío, o mediante una corriente de nitrógeno seco (pureza mínima 98 por 100). En todo caso, no debe calentarse por encima de 30 °C.
Hidrólisis por la lipasa pancreática. A 100 mg de la muestra de grasa, preparada como se ha indicado anteriormente (ver 34.5.5), pesados en el vasito descrito en 34.2.5, se añaden 20 miligramos de polvo de lipasa y 2 ml de la disolución 34.3.5. Se agita suavemente y se añaden 0,2 ml de disolución de cloruro cálcico al 22 por 100 y 0,5 ml de disolución de colato sódico al 0,1 por 100.
Se coloca el vasito, durante 1 minuto, en un baño a 40 °C. Se tapa y se agita fuertemente en el vibrador durante 2 minutos. Al cabo de este tiempo, se añade 1 ml de ácido clorhídrico 6N y 2 ml de éter etílico; se tapa y se agita con la mano. Se deja en reposo hasta que se separe la capa superior de éter etílico. Si se forma una emulsión, se rompe invirtiendo el tubo varias veces con suavidad.
Separación de los monoglicéridos por cromatografía en capa fina. Con ayuda de una jeringa adecuada, se coloca la mayor parte de la disolución etérea decantada en la parte inferior de una placa de gel de sílice de 20 x 20 cm, depositando las gotas de una forma regular en una línea horizontal situada a unos 15 mm del extremo inferior, dejando libre unos 10 mm en cada extremo. Una vez depositada la disolución, se introduce la placa en la cubeta, conteniendo el disolvente descrito en 34.3.11 en la cuantía necesaria para que la superficie quede unos 5 mm por debajo del producto depositado.
Cuando el frente del disolvente haya recorrido unos 15 centímetros, se saca la placa de la cubeta y se deja evaporar al aire el disolvente.
Observando la placa contra la luz de una ventana, se pueden ver perfectamente las bandas correspondientes a los productos formados en la liposis. La banda situada inmediatamente por encima de la posición original está constituida por los β-monoglicéridos. Con ayuda de una pequeña espátula, se raspa, recogiendo el polvo de sílice en un pequeño embudo de vástago ancho, colocado sobre el matraz de metilación (34.2.9).
Preparación de los ésteres metílicos Al matraz conteniendo la sílice raspada de la placa se agregan 10 ml de la disolución de metilato sódico (34.3.12), se coloca la varilla de reflujo, y se hierve durante 5 minutos.
Se añade una gota de fenolftaleína en metanol al 1 por 100 y, seguidamente, disolución de clorhídrico en metanol (34.3.15) gota a gota, hasta decoloración del indicador. Se hierve a re-flujo otros 5 minutos. Se deja enfriar añadiendo 1 ml de hexano o de éter de petróleo (34.3.10) y disolución acuosa saturada de cloruro sódico hasta que el hexano se sitúe en el cuello estrecho del matraz. Se agita fuertemente y se deja reposar. En pocos minutos se separa la disolución hexánica de los ésteres metílicos, que queda limpia y transparente en el cuello del matraz; pasándose, seguidamente, a inyectar esta disolución en el cromatógrafo para la determinación cualitativa y cuantitativa de los ácidos grasos.
Cromatografía en fase gaseosa de los ésteres metílicos. Los ésteres metílicos se determinan a continuación según el método número 41 ‒determinación de ácidos grasos por cromatografía en fase gaseosa‒ de los métodos oficiales de análisis de aceites y grasas.
34.5 Observaciones.
34.5.1 Si no se dispone de medidor de pH, puede utilizarse para su regulación en el medio reaccionante, el rojo cresol como indicador interno.
Para ello, al colocar el vaso en el termostato, según se indica en 34.4, se agregan 5 gotas de disolución acuosa al 1 por 100 del indicador (34.3.18) y se adiciona la disolución 0,1 N de hidróxido sódico, manteniendo una coloración rosa-violeta que corresponde, aproximadamente, a un pH de 8,3.
34.5.2 Con algunos aceites extraídos con disolventes, al efectuar la disolución en éter de petróleo o hexano puede formarse una turbidez que, si fuese muy intensa, podría perturbar el proceso cromatográfico de purificación. En estos casos, resulta aconsejable filtrar o centrifugar la disolución turbia, separando el sedimento y operando con la disolución transparente.
34.5.3 Si no se dispone de evaporador rotatorio, la eliminación del disolvente se puede realizar con análoga rapidez y eficacia, haciendo pasar por el interior del matraz una corriente de nitrógeno puro 98 por 100), calentando en baño de agua a temperatura que no exceda de 30 °C.
34.5.4 Teniendo en cuenta que se han tomado 20 g de muestra por cada 1 por 100 de acidez libre de oleico, se deberá adicionar 0,36 ml de la disolución de hidróxido sódico 2N. Si la fenolftaleína no acusase reacción alcalina, deberá adicionarse más disolución gota a gota, y agitando después de cada adición, hasta conseguir el viraje del indicador.
34.5.5 Con aceites comestibles de buena calidad aparente, y acidez reducida ≤3 por 100) en operaciones de rutina, se puede omitir el tratamiento previo de purificación con alúmina, ya que, en la inmensa mayoría de los casos, esta purificación no es necesaria. Sin embargo, como norma general, este tratamiento debe considerarse como necesario, y cualquier valor anormal obtenido efectuando una lipolisis directa no deberá ser aceptado sin confirmarlo previamente, con la grasa sometida al proceso de purificación.
34.6 Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.079.
35. RECONOCIMIENTO DEL ACEITE DE ORUJO DE ACEITUNA
(Prueba de Vizern)
35.1 Principio.
Este método tiene por objeto la identificación del aceite extraído por disolventes de los orujos grasos de aceituna.
Se aplica para determinar la pureza de los aceites de oliva vírgenes y refinados. En presencia de otros aceites, su aplicación e interpretación de resultados debe ser discrecional.
35.2 Material y aparatos.
35.2.(1, 2, 3 y 4) Como 22.2. (1, 2, 3 y 4).
35.2.5 Matraz erlenmeyer de 100 ml, con boca esmerilada, provisto de refrigerante de reflujo de aire, constituido por un tubo de 80 a 100 cm de longitud aproximadamente.
35.2.6 Tubo de ensayo de paredes gruesas, con dimensiones aproximadas de 160 mm de altura y 30 mm de diámetro interior.
35.2.7 Probeta de 10 ml.
35.3 Reactivos.
35.3.(1, 2, 3, 4, 5 y 6) Como 22.3. (1, 2, 3, 4, 5 y 6).
35.3.7 Alcohol etílico de 85 por 100 en volumen. Diluir 885 mililitros de etanol de 96° hasta 1.000 ml con agua destilada. La densidad será 0,8520. Comprobar con un alcohómetro.
35.4 Procedimiento.
Extraer el insaponificado de 5 g de aceite como en 22(a), empleando éter de petróleo.
La totalidad del insaponificable aislado recogido en el matraz de 100 ml se disuelve en 10 ml de etanol de 85°, en caliente con refrigerante de reflujo.
La disolución alcohólica se transvasa al tubo de ensayo 35.2.6 y se enjuaga el matraz con otros 10 ml de etanol de 85°, que se incorporan también al tubo de ensayo. Se homogeneiza la mezcla y se enfría a unos 25 °C; dejándolo en reposo durante una hora, a temperatura entre 20-25 °C. Observar al cabo de la hora.
La formación de un precipitado floconoso, que se deposita en el fondo del tubo, en forma de copos, acusa la presencia de aceite de orujo. Un precipitado más dividido o pulverulento, no resuelto en copos, no puede interpretarse como positivo, siendo muchos los aceites de oliva de presión que originan estos precipitados.
35.5 Expresión de los resultados.
Positivo o negativo.
35.6 Referencia.
1. M. Vizern. Ann. Fals. Fraud. 1953. 31.
36. TETRABROMUROS
(Prueba de Vizern-Guillot)
36.1 Principio.
Está fundamentada en la insolubilidad de los derivados tetrabromados de los ácidos grasos en éter de petróleo, en las condiciones especificadas en la prueba.
Se aplica al reconocimiento de aceites semisecantes en aceites de oliva.
La sensibilidad es del 5 por 100 para el aceite de soja y del 10 por 100 para los demás aceites.
36.2 Material y aparatos.
36.2.1 Tubo de vidrio con tapón esmerilado de aproximadamente 20 mm de diámetro y 180 mm de largo.
36.3 Reactivos.
36.3.1 Éter de petróleo (p. e. 40-70 °C) exento de productos reaccionables con los halógenos.
36.3.2 Bromo para análisis, exento de cloro.
36.3.3 Reactivo de bromación. Agregar gota a gota y agitando continuamente 4 ml de bromo a 100 ml de éter de petróleo, enfriado previamente a 0 °C. Conservar el reactivo en hielo fundente hasta el momento del ensayo.
36.4 Procedimiento.
Filtrar el aceite a ensayar que debe estar totalmente exento de humedad.
En el tubo de ensayo, desecado, introducir 1 ml de aceite seco y filtrado, disolverlo en 10 ml de éter de petróleo. Mantenerlo durante 5 minutos en hielo fundente, con el tubo cerrado. Agregar en pequeñas adiciones 10 ml de reactivo de bromación, agitar ligeramente y mantener el conjunto durante una hora en hielo fundente, al cabo de dicho tiempo el color de la disolución debe indicar la permanencia de exceso de bromo.
Observar el aspecto de la disolución. La presencia de aceite semisecante se acusa por la formación de un precipitado más o menos intenso, según la composición del aceite y su contenido en la mezcla, los aceites de oliva puros darán un ensayo limpio y transparente.
36.5 Expresión de los resultados.
Positivo o negativo.
36.6 Referencia.
1. M. Vizern. Ann. Fals. Fraud. 1953. 31.
37. MODIFICACIÓN SYNODINOS-KONSTAS
(Prueba de Hauchecorne)
37.1 Principio.
Se hace actuar el ácido nítrico concentrado sobre el aceite, el cual reacciona con productos de oxidación originados en las materias grasas parcialmente alteradas, hayan sido sometidas o no a un proceso de transformación industrial.
Es aplicable solamente a aceites de oliva «vírgenes» y refinados o a sus mezclas, pudiéndose obtener una coloración no satisfactoria en aceites no adulterados, cuya calidad deficiente o su conservación inadecuada sea responsable de la respuesta obtenida en el ensayo.
37.2 Material y aparatos.
37.2.1 Frascos de unos 50 ml de capacidad con tapón esmerilado
37.2.2 Probeta graduada con tapón esmerilado de 25 ml.
37.2.3 Embudo de 6 cm de diámetro.
37.2.4 Probeta graduada de 30 ml.
37.2.5 Papel de filtro corriente.
37.3 Reactivos.
37.3.1 Tierra decolorante para aceites de elevada actividad.
37.3.2 Ácido nítrico, para análisis, densidad 1,40.
37.4 Procedimiento.
Medir, con la probeta graduada, 30 ml de aceite de la muestra y verter en el frasco de 50 ml. Pesar 3 g de tierra decolorante y agregarlos al frasco, agitándose fuertemente durante 30 segundos.
El producto tratado con tierra se filtra, por filtro de pliegues, recogiéndose 10 ml en la probeta de tapón. Si se dispusiere de centrífuga, sería preferible centrifugar realizándose la operación en menos tiempo y con más eficacia.
Agregar, al aceite contenido en la probeta, 10 ml de ácido nítrico (d = 1,40) y agitar durante unos 30 segundos, dejándose reposar seguidamente y observándose el color desarrollado en la capa superior oleosa (ver 4.6).
37.5 Expresión de los resultados.
El aceite de oliva «virgen» de características normales y bien conservado, produce una coloración amarilla clara.
Los aceites de oliva refinados, procedentes de un «lampante» de buena calidad, suelen dar un color amarillo sucio que, frecuentemente, se acerca al amarillo tostado.
Los aceites de orujo refinados y, en general, los aceites de calidad deficiente, tanto «vírgenes» como refinados o las mezclas de ambos, dan una coloración marrón, obteniéndose una respuesta análoga con los aceites esterificados.
Los aceites de semillas refinados dan coloraciones muy diversas que no pueden considerarse como específicas para cada uno, pero que, sin embargo, un operador experto en la aplicación de la prueba los diferencia perfectamente de las coloraciones atribuibles a los aceites de oliva.
37.6 Observaciones.
37.6.1 El tiempo de desarrollo de estas coloraciones es muy variable. Generalmente se puede observar a los 2 ó 3 minutos de haberse separado la capa oleosa de la capa ácida, pero no se debe dar el dictamen definitivo hasta observar la coloración transcurridos unos 15 minutos de reposo.
37.7 Referencia.
1 Instituto de Racionalización del Trabajo. Una Norma Española 55.053.
38. RECONOCIMIENTO DEL ACEITE DE ALGODÓN
(Reacción de Halphen)
38.1 Principio.
Este método detecta cualitativamente el aceite de algodón en mezcla con otros aceites o grasas vegetales o animales.
Puede considerarse específica del aceite de algodón, excepto en presencia de aceites de Kapok y de grasas procedentes de animales que hayan sido alimentados con harina de semilla de algodón o alguna materia que contenga ácidos ciclopropenoides; en ambos casos habrá que realizar pruebas discriminatorias.
38.2 Material y aparatos.
38.2.1 Tubos de ensayo de dimensiones 250 x 25 mm aproximadamente.
38.2.2 Baño de agua caliente con temperatura regulable
38.2.3 Baño de aceite o bloque de calefacción adecuado para los tubos de ensayo que han de ser utilizados, regulables a una temperatura de 110-115 °C. En lugar de aceite puede emplearse una disolución acuosa saturada de cloruro sódico.
38.3 Reactivos.
38.3.1 Preparar una disolución de azufre en bisulfuro de carbono al 1 por 100 y añadir un volumen igual de alcohol amílico.
38.4 Procedimiento.
Tomar en un tubo de ensayo de 10 ml de aceite muestra y agregar un volumen igual de reactivo. Agitar y calentar en un baño de agua a 70-80 °C durante unos cuantos minutos, agitando de cuando en cuando, hasta que el bisulfuro de carbono comience a hervir y la muestra comience a desprender vapores.
Colocar el tubo en un baño regulado a 110-115 °C durante 1-2 horas. La presencia de aceite de algodón se acusa por el desarrollo de un color rojo más o menos intenso; los aceites refinados suelen dar color rosa. En el caso de débiles contenidos de aceite de algodón por bajo del límite de sensibilidad del ensayo, el líquido toma color amarillo.
38.5 Expresión de resultados.
Positiva o negativa.
38.6 Observaciones.
38.6.1 La estructura del ciclopropeno responsable del desarrollo del color se destruye por calefacción, y a 200 °C la destrucción es prácticamente completa. Por tanto, los aceites refinados suelen dar, a lo sumo, una reacción débil y frecuentemente dan resultado negativo. Del mismo modo, la hidrogenación y los tratamientos con ácidos u otros agentes químicos reducen la intensidad y pueden anularla por completo.
38.6.2 Diferentes lotes de aceite de algodón pueden reaccionar con intensidades diferentes. La intensidad del color no puede tomarse como índice del contenido en aceite de algodón.
38.7 Referencias.
1. J. Pharm. Chim. 1897. 6, 390.
2. Analyst. 1897. 22, 396
3. American Oil Chemists' Society. Official and Tentative Methods. Cb. 1-25.
39. RECONOCIMIENTO DEL ACEITE DE SEMILLA DE TÉ EN ACEITE DE OLIVA
(Reacción de Fitelson)
39.1 Principio.
Este método, es aplicable solamente a la identificación de aceite de semilla de té en mezcla con aceite de oliva.
39.2 Material y aparatos.
39.2.1 Tubos de ensayo con dimensiones aproximadas de 150 × 15 mm.
39.2.2 Pipeta de 1,5 ml, graduada en décimas.
39.2.3 Pipeta cuentagotas, con orificio de salida de, aproximadamente, 2 mm de diámetro, calibrado de tal forma que siete gotas de aceite pesen, aproximadamente, 0,22 g.
39.3 Reactivos.
39.3.1 Cloroformo, reactivo análisis.
39.3.2 Acido sulfúrico, reactivo análisis, d = 1,84.
39.3.3 Anhídrido acético, reactivo análisis.
39.3.4 Éter etílico anhidro, reactivo análisis.
39.4 Procedimiento.
Colocar en un tubo de ensayo, medidos con pipeta, 0,8 ml de anhídrido acético, 1,5 ml de cloroformo y 0,2 ml de ácido sulfúrico. Mezclar y enfriar en un baño de agua con hielo a °C.
Añadir con la pipeta cuentagotas 0,22 g de la muestra de aceite. Si aparece turbidez, añadir anhídrido acético, gota a gota, agitando después de cada adición hasta que desaparezca. Mantener a 5 °C durante 5 minutos.
Añadir 10 ml de éter etílico, previamente enfriado a 5 °C, tapar el tubo de ensayo con un corcho y mezclar inmediatamente invirtiendo el tubo dos veces. Colocar nuevamente el tubo en el baño de agua-hielo y observar el color.
El aceite de semilla de té puro da una coloración roja intensa, alcanza un máximo y seguidamente desaparece.
39.5 Expresión de resultados.
Positiva o negativa.
39.6 Observaciones.
39.6.1 Muchas muestras genuinas de aceite de oliva dan coloraciones rosa de una intensidad comparable a un contenido de aceite de semilla de té del 10 por 100 o más. Por consiguiente, coloraciones rosadas débiles en muestras presentadas como aceite de oliva puro no pueden ser atribuidas a la presencia de aceite de semilla de té.
39.6.2 Algunas muestras de aceite de oliva, especialmente de calidad refinable o tipos industriales muestran una coloración oscura que tiende a enmascarar el color rojo característico de la reacción. Para evitar este inconveniente, proceder como sigue: Saponificar una mezcla de 5 g de la muestra y 5 g de parafina líquida blanca, utilizando un exceso de hidróxido potásico en disolución alcohólica (aproximadamente 5 ml de hidróxido potásico al 50 por 100 diluido con 30 ml de alcohol etílico). Una ebullición mantenida durante 10-15 minutos es generalmente suficiente. Pasar la disolución del producto saponificado a una ampolla de decantación y añadir un volumen igual de agua destilada, mezclar y dejar que decante, separándose en dos capas. Separar la capa inferior constituida por la disolución jabonosa. Lavar la capa oleosa varias veces con agua destilada con el fin de eliminar los restos de jabón. Secar el producto una vez lavado, añadiendo sulfato sódico anhidro y dejar en reposo durante unos minutos. Filtrar y operar como se indica en 39.4.
39.6.3 Es aconsejable trabajar simultáneamente y en forma idéntica con muestras especialmente preparadas que sirvan como patrones de comparación.
39.7 Referencias.
1. J. Assoc. Offic. Agr. Chem. 1936. 19, 493-97.
2. American Oil Chemists' Society. Official and Tentative Methods. Cb. 3-39.
40. RECONOCIMIENTO DEL ACEITE DE SÉSAMO
(Reacción de Pavolini)
40.1 Principio.
Este método tiene por objeto el reconocimiento del aceite de sésamo en mezcla con otros aceites y grasas comestibles.
Es aplicable aunque el aceite de sésamo se haya sometido a un proceso de refinación alcalina, o hidrogenación parcial para la preparación de «shortenings» o margarinas.
40.2 Material y aparatos.
40.2.1 Tubo de ensayo de 20 × 110 mm de diámetro interior y longitud, respectivamente, con tapón esmerilado y llave de salida de vidrio en su parte inferior. Debe llevar una marca indicando los volúmenes de 10 ml y 15 ml.
40.2.2 Soporte para mantener el tubo anterior en posición vertical.
40.2.3 Cápsula de porcelana, fondo plano, de unos 60 mm de diámetro.
40.3 Reactivos.
40.3.1 Acido sulfúrico concentrado (d = 1,84).
40.3.2 Disolución de 0,35 ml de furfurol, recientemente destilado, en un litro de anhídrido acético. Esta disolución debe guardarse en frascos oscuros, de tapón esmerilado, preferiblemente provistos de capuchón protector, también con ajuste esmerilado. Manteniendo en debidas condiciones el reactivo, se conserva largo tiempo. No es necesario que el anhídrido ascético sea rigurosamente anhidro, pudiéndose tolerar hasta un 5 por 100 de agua.
40.4 Procedimiento.
En el tubo graduado se vierten 10 ml de la muestra de aceite, y seguidamente 5 ml de reactivo 40.3.2. Agitar durante 1 minuto aproximadamente, colocar el tubo en el soporte y dejar reposar hasta que se separen las dos capas.
Verter de 1 a 2 ml de la capa inferior a la cápsula de porcelana. Agregar 5 ó 8 gotas de ácido sulfúrico concentrado y mezclar imprimiendo a la cápsula un suave movimiento de rotación.
Para cantidades de aceite de sésamo superiores al 1 por 100 se desarrolla inmediatamente una coloración azul de Prusia oscura, con ligero tinte verdoso. Para contenidos menores, el color azul va siendo más débil, aumentando al mismo tiempo el período requerido para el desarrollo completo del color. En mezcla con aceites de oliva y soja, el límite de detección es de 0,25 por 100, acusándose la presencia por la aparición de un color gris azulado, que tarda en desarrollarse de 5 a 10 minutos.
40.5 Expresión de resultados.
Positivo o negativo.
40.6 Referencias.
1. Pavolini L. Olii Minerali, Grassi Saponi, Colori Vernici; 12, 41, 1934.
2. Instituto de Racionalización del Trabajo. Una Norma Española 55.068.
41. DETERMINACIÓN DE ÁCIDOS GRASOS POR CROMATOGRAFÍA EN FASE GASEOSA
41.1 Principio.
El método está basado en la separación y determinación cromatográfica de los ésteres metílicos de los ácidos grasos.
Es aplicable a las grasas vegetales y animales con ácidos grasos de 12 a 24 átomos de carbono No es aplicable para la determinación de ácidos grasos oxidados y epoxiácidos.
41.2 Material y aparatos.
41.2.1 Matraz redondo, fondo plano, de 100 ml, boca esmerilada.
41.2.2 Refrigerante de reflujo, adaptable al matraz anterior.
41.2.3 Ampollas de decantación de 500 ml.
41.2.4 Matraz redondo, fondo plano, de 200 ml.
41.2.5 Cromatógrafo apto para separar los esteres metílicos, provisto de horno con regulación de temperatura hasta 250-300 °C, e inyector susceptible de ser calentado hasta 50 °C por encima de la temperatura del horno. Detector suficientemente sensible y aparato registrador continuo.
41.2.6 Botella con nitrógeno a presión; riqueza mínima 99,8 por 100.
41.2.7 Columna de acero inoxidable, aluminio, cobre o vidrio de 3-6 mm de diámetro interior, de 2 m de longitud, rellena con Chromosorb P o W (60-80 mallas) o Celite 545 (80-100 mallas), impregnada con 5 por 100 de succinato de polietilenglicol.
41.2.8 Jeringa, de 5 a 10 μl de capacidad.
41.3 Reactivos.
41.3.1 Metanol absoluto, reactivo para análisis.
41.3.2 Sodio metálico, reactivo para análisis.
41.3.3 Disolución de metilato sódico. Disolver 5 g de sodio metal en 1.000 ml de metanol absoluto (aprox. 0,2 N).
41.3.4 Éter de petróleo (p. e., 40°-60 °C) o hexano, para cromatografía.
41.3.5 Disolución de ácido clorhídrico anhidro en metanol absoluto al 3-4 por 100.
41.3.6 Cloruro sódico, reactivo para análisis.
41.3.7 Sulfato sódico, reactivo para análisis.
41.3.8 Disolución de fenolftaleína al 1 por 100 en metanol.
41.3.9 Disolución de rojo de metilo al 0,1 por 100 en etanol al 60 por 100
41.3.10 Nitrógeno gas puro (99,8 por 100).
41.4 Procedimiento.
41.4.1 Preparación de los ésteres metílicos.
41.4.1.1 Aceites y grasas de cualquier acidez e insaponificable no superior al 2 por 100.
a) Pesar 1 g de grasa en matraz de 100 ml. Agregar 25 ml de disolución de metilato sódico. Adaptar refrigerante de reflujo y hervir hasta obtener una sola fase; aproximadamente, 5 minutos. Interrumpir la calefacción.
b) Agregar al matraz 30 ml de disolución de ácido clorhídrico en metanol. Volver a calentar a reflujo 5 minutos. Enfriar. c/ Extracción de los ésteres metílicos.
Método rápido. Pasar la disolución obtenida en el matraz a otro de 100 ml con cuello estrecho y largo. Lavar el primero con 4-5 ml de hexano o heptano y pasarlos al segundo; calentar éste sin llegar a hervir, imprimiéndole movimiento de rotación durante 1 ó 2 minutos. Agregar disolución saturada de cloruro sódico en cantidad suficiente para situar la capa de hexano en el cuello del matraz; dicha capa presentará aspecto limpio y transparente. Tomar con una pipeta 2 ó 3 ml, pasarlos a un recipiente adecuado para su conservación o inyección directa en el cromatógrafo.
Método de referencia. Pasar la disolución contenida en el matraz a una ampolla de extracción de 500 ml, lavar aquél con unos 30 ml de éter de petróleo o hexano. Añadir a la ampolla 123 ml de agua destilada. Agitar fuertemente y dejar reposar. Extraer tres veces consecutivas la capa inferior con 30 ml de éter o hexano cada vez. Lavar repetidamente los tres extractos reunida: en una ampolla de extracción con 15 ml de agua des-rada cada vez, hasta eliminar completamente el ácido, empleando rojo de metilo como indicador. Secar la disolución con sulfato sódico anhidro Eliminar el disolvente en bario de agua bajo corriente de nitrógeno.
41.4.1.2 Aceites y grasas de acidez no superior al 0,3 por 100 en ácido oleico.
Omitir la metanolisis en medio ácido. Realizar la metanolisis alcalina según 41.4.1.1.a) y, estando aún caliente el matraz, agregar por la parte superior del refrigerante una gota de disolución de fenolftaleína y disolución de ácido clorhídrico en metanol en cantidad algo superior a la necesaria para neutralizar el metilado. Dejar enfriar. Pasar la disolución a la ampolla de extracción. Proceder a la extracción de los ésteres metílicos según 41.4.1.1.c).
41.4.1.3 Aceites y grasas de acidez superior al 80 por 100 en ácido oleico
Omitir la metanolisis alcalina. Realizar la metanolisis ácida, pesando en el matraz 2 g de muestra, previamente filtrada y seca. Agregar 60 ml de la disolución de ácido clorhídrico en metanol. Proseguir según 41.4.1.1.b) y 41.4.1.1.c).
41.4.1.4 Aceites y grasas con insaponificable superior al 2 por 100.
Eliminar la materia insaponificable según el método «Determinación de la materia insaponificable». La solución hidroalcohólica de jabones resultantes se concentra en evaporador rotario hasta unos 50 ml. Pasar éstos a una ampolla de extracción. Acidificar con ácido clorhídrico 2 N hasta reacción ácida al rojo de metilo. Extraer con éter de petróleo o hexano. Proseguir como en 41 4 1 3.
41.4.2 Preparación de la columna.
41.4.2.1 Soporte: En un vaso de 600 ml colocar 100 g de Chromosorb W o P, o Celita 545, preferentemente el primero, agregar ácido clorhídrico al 20 por 100 en cantidad suficiente hasta cubrir el producto. Dejar en reposo durante 15 horas. Decantar el ácido. Agregar agua destilada, agitando fuertemente, y dejar reposar 5 minutos. Decantar el agua y volver a cubrir con ácido clorhídrico al 20 por 100 y reposar durante 2 horas. Filtrar sobre embudo de vidrio y lavar con agua destilada hasta neutralidad. Extender seguidamente el soporte en capa delgada en cápsulas de porcelana o cubeta de vidrio. Desecar en estufa a 110 °C durante 2 horas.
41.4.2.2 Fase estacionaria: Succinato de polietilenglicol comercial.
41.4.2.3 Impregnación del soporte: Disolver, en cápsula de porcelana, 0,5 g de la fase estacionaria o fija de unos 50 ml de acetona o cloroformo. Añadir 9,5 g de soporte tratado. Regular el disolvente para obtener una papilla de fluidez adecuada para una buena homogeneización por agitación. Eliminar el disolvente sobre baño de agua, removiendo con espátula, hasta obtener el producto en forma de polvo suelto. Extenderlo en capa delgada e introducirlo en estufa a 100 °C durante 2 horas.
41.4.2.4 Llenado de la columna: Introducir el soporte impregnado con fase fija en la columna, haciéndola vibrar por percusión o con un vibrador magnético, hasta su llenado total. Es preciso que el material rellene la columna homogéneamente, evitando la formación de vías o canales, para que no pierda eficacia. Obturar los extremos con tapón de lana de vidrio y/o cilindros de malla metálica de cobre.
41.4.3 Condiciones operatorias.
Acoplar al cromatógrafo la columna preparada, o la suministrada por la firma comercial del aparato.
Regular la temperatura del horno entre 160-170 °C, la cámara de inyección 50 °C más alta que la del horno y el detector 25 °C sobre la de la columna.
Ajustar el flujo de gas portador, en principio del orden de 4 l/hora, y mantenerlo constante. Acondicionar la nueva columna para la temperatura de operación, fluyendo gas portador durante 24 horas o hasta estabilidad. Registrar la línea base para comprobar la estabilidad del aparato.
Operar con patrones, rectificando las condiciones operatorias a la vista de los resultados obtenidos, regulando el flujo de gas portador de forma que el linolenato de metilo sea eluido en 30 minutos, como máximo. Medirlo periódicamente con el medidor de flujo de burbuja de jabón y otro medio.
Preparar una disolución de los ésteres metílicos en acetona o hexano de pureza comprobada. Inyectar de 0,4 a 0,6 μl, con jeringa, descargando y retirando la aguja rápidamente.
El registro obtenido será admisible si cumple las siguientes condiciones: a) El área total de los picos registrados, referida a la sensibilidad máxima utilizada, no será inferior a 5.000 mm2. De esta forma, los componentes presentes en la cuantía de 0,2 por 100 deberán dar picos de 10 mm2, como mínimo. b) Todos los picos han de caer dentro del papel registrador.
Señalar en el cromatograma el punto de referencia de introducción de la muestra, los picos iniciales de salida del aire y la atenuación de registro de cada pico.
41.4.4 Comprobación de las condiciones de trabajo del instrumento y de la columna.
Se realiza cromatografiando una muestra de patrones conteniendo aproximadamente cantidades iguales de estearato y oleato de metilo, inyectando una cantidad de muestra tal que los picos registrados sean del 25 al 50 por 100 del ancho del papel de registro, y determinando la resolución de los dos ácidos según:

D = distancia entre los dos máximos de los picos del oleato estearato.
O = ancho de la base del oleato.
E = ancho de la base del estearato.
Si la resolución es igual o mayor de 1,0, el instrumento y columna están en condiciones de trabajo satisfactorias.
Todas las columnas experimentan con el uso una pérdida gradual de resolución; cuando ésta es menor de 1,0, la columna será reemplazada.
41.5 Cálculo.
41.5.1 Identificación de los picos. Los ésteres aparecen en orden creciente del número de átomos de carbono y de aumento de insaturación para un mismo número de átomos de carbono. El palmítico (C16) aparece delante de los C18, y éstos en el orden oleato (C18 : 1), linoleato (C18 : 2) y linoleato (C18 : 3); el aráquico (C20 : 0) aparece normalmente después del (C18 : 3), pero pueden estar invertidos en algunos casos. Se establecerá la identidad para una columna dada inyectando muestras patrones.
Fijar los tiempos de retención, medidos en el cromatograma (distancia entre el máximo de cada pico y la posición del pico inicial de salida del aire, o del pico de salida del disolvente en el caso de detectores de llama de hidrógeno). Compararlos con los obtenidos con las mezclas patrones.
41.5.2 Determinación cuantitativa. Se considera que la superficie del triángulo formado por cada pico y la línea base es proporcional a la cantidad de componente identificado con dicho pico.
Determinar el área trazando líneas tangentes a los lados de cada pico prolongadas hasta cortar la línea base. Multiplicar la altura del triángulo (corregida para la atenuación empleada) por la mitad de la base.
Si se ha registrado con atenuación única, medir el área multiplicando la altura de] pico por su ancho a la mitad de la altura.
Sumar las áreas de todos los picos y calcular el tanto por ciento correspondiente a cada pico:

Pi = tanto por ciento de cada componente en la mezcla.
A1 = área de cada pico.
Normalmente se empleará este criterio y expresión.
41.5.2.1 Factores de corrección. En algunos casos, por no ser lineal la respuesta del instrumento y por las diferencias en los pesos moleculares, se determinan los factores de corrección de cada ácido, referidos al ácido palmítico, analizando mezclas conocidas de composición similar a la mezcla problema.

fx = factor de corrección del ácido.
x = tanto por ciento del ácido en la mezcla.
Ax = área del pico x.
P = tanto por ciento del ácido palmítico en la mezcla patrón.
Ap = área del pico del ácido palmítico.

41.6 Referencias.
1. Jacini y otros. La Rivista Italiana de la Sostanze Grasse. 1963. 108, 40.
2. Firestone, D. Journal Association of Official Agricultura] Chemists. 1963. 46, 1.
3. International Union of Pure and Applied Chemistry Standard Methods for the Analysis of Oils, Fats and Soaps. 1964. II. D. 19. Suplemento 5.° ed.
4. Consejo Oleícola Internacional. Métodos de Análisis. 1967.
42. DETERMINACIÓN DEL CONTENIDO EN MATERIA GRASA DE LOS ALPECHINES
42.1 Principio.
Se considera materia grasa del alpechín el producto obtenido por extracción con éter de petróleo en condiciones determinadas.
El procedimiento es aplicable a toda clase de alpechines, ya sean centrifugados o no.
42.2 Material y aparatos.
42.2.1 Ampollas de extracción de un litro y litro y medio de capacidad.
42.2.2 Vaso de precipitado de un litro y medio de capacidad.
42.2.3 Matraz redondo, de fondo plano, de 250 ml de capacidad.
42.2.4 Embudo de pico corto de 80 mm de diámetro.
42.3 Reactivos.
42.3.1 Éter de petróleo, para análisis (ver 42.6.1).
42.3.2 Sulfato sódico anhidro puro. 50 gr de este producto, introducido en un cartucho y extraído en un Sohxlet con éter de petróleo durante cinco horas, no debe dejar en el matraz, una vez desecado en estufa a 105°C durante dos horas, un residuo superior a un centigramo.
42.4 Procedimiento.
Pesar, en el vaso de precipitado de la muestra bien homogeneizada por agitación, un kilogramo de alpechín (42.6.2.).
Trasvasar a la ampolla de extracción de un litro y medio, lavando con pequeñas cantidades de agua destilada, con objeto de arrastrar los restos de alpechín que puedan quedar en el vaso. Medir 150 ml de éter de petróleo y verterlos en el vaso de precipitado, cuidando de que lave bien las paredes y fondo del vaso. Pasar a la ampolla de extracción y agitar fuertemente, tomando las precauciones necesarias. Dejar reposar el tiempo suficiente para que se separen perfectamente la capa de éter de petróleo del alpechín (42.3.3).
Pasar la capa de alpechín al vaso de precipitado. Trasvasar la solución de éter de petróleo por la parte superior de la ampolla de extracción a una segunda ampolla de un litro de capacidad, en la que se han introducido previamente unos 200 ml de agua destilada, cuidando de no arrastrar restos de alpechín.
Repetir la extracción del alpechín dos veces más, como mínimo (ver 42.6.4). Las tres fracciones de éter de petróleo reunidas en la segunda ampolla se lavan con agua destilada por agitación.
Repetir los lavados, si es necesario, hasta que el agua destilada decantada no presente coloración.
Una vez separada el agua del último lavado, se añade una pequeña cantidad de sulfato sódico anhidro a la solución de éter de petróleo, agitando y dejando reposar media hora, como mínimo. Se filtra sobre el matraz de 250 ml, previamente mantenido dos horas en estufa a 105 °C, enfriado en desecador y tarado, hasta ocupar la mitad del matraz; se destila el éter volviendo a filtrar otra porción de éter de petróleo, que se destila seguidamente, repitiendo sucesivamente estas operaciones hasta agotar la totalidad existente en la ampolla, que se enjuaga con 20ó 25 ml de éter de petróleo, que se adicionan también al matraz.
Una vez terminada la destilación, se introduce el matraz en una estufa regulada a 105 °C, en la que se mantiene durante dos horas. Dejar enfriar en un desecador y pesar con precisión de miligramo.
42.5 Cálculos.
El tanto por ciento de grasa se obtendrá mediante la fórmula:

en la que:
P2 = peso en gramos del matraz con el aceite.
P1 = peso en gramos del matraz vacío y desecado.
P0 = peso en gramos de la muestra de alpechín.
42.6.1 Se puede sustituir el éter de petróleo, sin variación sensible en los resultados, por el hexano técnico.
42.6.2 En algunas muestras de alpechín, especialmente los de elevado contenido graso, resulta prácticamente imposible conseguir una homogeneización adecuada por simple agitación en el recipiente, En estos casos es aconsejable agregar una cantidad aproximada de 0,1 g de «Tween 80» por litro de muestra, provocándose una emulsión que facilita la homogeneización y toma de una muestra representativa, sin afectar prácticamente los resultados cuantitativos.
42.6.3 Las emulsiones persistentes que puedan originarse ocasionalmente pueden hacerse desaparecer añadiendo pequeñas cantidades de alcohol etílico.
42.6.4 En alpechines ricos en materia grasa, es aconsejable realizar mayor número de extracciones.
42.7 Referencia.
1. Instituto de Racionalización del Trabajo. Una Norma Española 55.114.
1. DETERMINACIÓN DEL ÍNDICE DE MATERIAS CELULÓSICAS
1.1 Principio.
Las «materias celulósicas» representan las sustancias no digestibles de origen vegetal que constituyen el residuo que queda después de un ataque ácido en condiciones bien definidas, con una mezcla de ácido acético, ácido nítrico y ácido tricloroacético. Después de hervir la muestra con la mezcla de ácidos se separa el residuo insoluble, se seca y se incinera.
El índice de materias celulósicas se calcula mediante la pérdida de masa en el curso de la incineración.
La aproximación debe ser del 0,1 por 100.
1.2 Material y aparatos.
1.2.1 Molino de laboratorio (se puede emplear cualquier tipo, excepto de bolas).
1.2.2 Matraces cónicos erlenmeyer de 200 ó 300 ml de boca normalizada.
1.2.3 Refrigerador de reflujo, de boca normalizada.
1.2.4 Mechero Bunsen.
1.2.5 Tela metálica con amianto o placa de material refractario.
1.2.6 Soporte de trípode o aparato análogo.
1.2.7 Matraz de vacío con tulipa (Kitasato).
1.2.8 Agitadores de vidrio. Un agitador de vidrio revestido de caucho en la extremidad.
1.2.9 Crisol filtrante de cuarzo o de vidrio (porosidad de 40 a 90 mm).
1.2.10 Placas filtrantes de porcelana que cubran totalmente la superficie filtrante.
1.2.11 Trompa de agua.
1.2.12 Desecador conteniendo silicagel coloreado en azul (diámetro aproximado 25 cm).
1.2.13 Balanza de precisión (sensibilidad 0,1 mg).
1.2.14 Estufa.
1.2.15 Horno eléctrico para incineración.
1.2.16 Placa de material refractario.
1.2.17 Cartuchos deshidratantes de silicagel coloreado de azul.
1.3 Reactivos.
1.3.1 Acido acético al 70 por 100 d. a 20 °C = 1,07 (730 g de ácido acético glacial del 96 por 100 diluido hasta 1.000 g con agua destilada).
1.3.2 Acido nítrico concentrado, d. a 20 °C = 1,4.
1.3.3 Acido tricloroacético, p.p.a. cristalizado.
1.3.4 Acetona pura.
1.3.5 Éter etílico puro.
1.3.6 Arena de mar, diámetro inferior a 0,5 mm.
1.3.7 Fragmentos de tierra cocida (platos porosos machacados), diámetro de 0,5 a 2 mm.
1.4 Procedimiento.
1.4.1 Preparación de la solución ácida para la hidrólisis: Mezclar 900 ml de ácido acético con 60 ml de ácido nítrico y 24 g de ácido tricloroacético (d. 20 °C = 1,10).
1.4.2 Preparación de la arena de mar y de los crisoles.
Hacer hervir la arena de mar con ácido clorhídrico (4N) para eliminar el hierro, lavar con agua destilada para eliminar el cloruro y calcinar a 550 °C durante seis horas. Preparar las placas de porcelana y el polvo de tierra cocida de la misma forma.
Antes de la primera utilización de los crisoles filtrantes de cuarzo o vidrio, limpiarlos cuidadosamente e incinerarlos durante seis horas a 550 °C. Para evitar tensiones en la parte inferior deslustrada, colocar los crisoles filtrantes de vidrio en el horno de incineración frío y no retirarlos hasta después de enfriarlo alrededor de 200 °C. Los crisoles de cuarzo no experimentan tensiones y se pueden poner o sacar con toda seguridad en el horno caliente.
Introducir en los crisoles filtrantes de 5 a 6 g de arena de mar. Igualar la superficie. Introducir a continuación de 4 a 5 g de polvo de tierra cocida e igualar del mismo modo la superficie. Colocar a continuación la placa filtrante de porcelana encima de estas dos capas y apretar ligeramente. Se puede utilizar de nuevo el crisol así preparado, sin limpieza especial, pero es necesario verificar la permanencia de las capas.
1.4.3 Preparación de las muestras a ensayar.
Triturar la muestra de forma que el 95 por 100 del producto pase a través de un tamiz de 1,0 mm. Solamente para algunas sustancias fibrosas, tales como los granos de avena, no se alcanza tal grado de finura.
Está prohibido el empleo de molinos de bolas.
1.4.4 Cantidad inicial de la muestra a ensayar.
La cantidad de muestra a tomar depende del contenido en materias celulósicas del producto de forma que se obtenga finalmente una masa de materias celulósicas entre 50 y 150 mg. Para un producto que contenga materias celulósicas en cantidad inferior al 5 por 100, tomar 3 g de sustancia y utilizar 60 ml de solución ácida; para un contenido en materias celulósicas elevado, hacer hervir 102 g de materia en 40 ml de mezcla disolvente.
1.4.5 Dosificación.
Pesar la muestra a ensayar triturada y ponerla en suspensión en el matraz cónico erlenmeyer con un tercio de la solución ácida que debe corresponder a 20 veces el peso de la muestra. Deshacer los grumos con un agitador de vidrio que debe permanecer en el erlenmeyer. Lavar cuidadosamente la pared del erlenmeyer con el resto de la solución ácida para que no quede ninguna partícula de la sustancia adherida a la pared. Para evitar que la sustancia suba a lo largo de las paredes, acoplar cuidadosamente el erlenmeyer con el refrigerador de reflujo. Calentar de forma que se alcance la temperatura de ebullición en tres minutos. Regular la llama del mechero Bunsen para que la altura de la espuma formada no sobrepase 10 mm. Mantener la ebullición de la muestra durante 30 minutos exactamente, sin agitar el frasco ni la suspensión.
Filtrar a continuación bajo vacío la suspensión en ebullición con ayuda de una trompa de agua y a través del crisol filtrante preparado. Regular el vacío para asegurar la filtración continua. Lavar el erlenmeyer y el agitador de vidrio con agua destilada y caliente (de 70 a 80 °C) y trasvasar totalmente los residuos de materias celulósicas en el crisol filtrante con ayuda de un agitador revestido de caucho. Son necesarios, para un lavado cuidadoso y rápido, de 300 a 400 ml de agua destilada y caliente para obtener la reacción neutra (comprobar con papel tornasol en el agua de lavado filtrada).
Inmediatamente después del lavado, vaciar el matraz de vacío. Llenar el crisol filtrante tres veces con acetona y dejar filtrar sin ayuda del vacío (pero si el filtrado es demasiado lento, aspirar ligeramente sin sobrepasar la velocidad de una gota por segundo). Lavar a continuación dos veces con éter etílico y aspirar fuertemente los vapores residuales de éter. Secar previamente los crisoles filtrantes durante algunos minutos y pesarlos aproximadamente.
Secar los crisoles filtrantes en una estufa calentada a 130 °C durante 60 minutos exactos, introducir a continuación cuatro crisoles como máximo en un desecador y dejarlos enfriar algunos minutos antes de poner la tapa. Dejar enfriar el desecador cerrado durante una hora al lado de la balanza. En el transcurso del enfriamiento y de la pesada, colocar dos cartuchos deshidratantes de silicagel en la balanza de precisión. Pesar rápidamente (es por lo que hay que conocer previamente el peso de los crisoles llenos antes del secado).
Calcinar los crisoles filtrantes de cuarzo en el horno eléctrico previamente calentado a 550 °C, durante 30 minutos, colocarlos a continuación sobre una placa refractaria durante cinco minutos por lo menos, para que se enfríen hasta 100 °C, aproximadamente. Introducir de nuevo cuatro crisoles como máximo en un desecador, dejándolos enfriar al lado de la balanza durante una hora exactamente y pesar rápidamente.
Si se emplean crisoles filtrantes de vidrio, ponerlos en el horno frío, a fin de evitar las tensiones en la parte inferior deslustrada. Después de alcanzar la temperatura de 550 °C, incinerar durante treinta minutos, dejar enfriar los crisoles en el horno abierto hasta 200 °C, colocarlos a continuación algunos minutos sobre la placa refractaria para dejarlos enfriar hasta 100 °C, aproximadamente, y operar después como para los crisoles filtrantes de cuarzo.
No es necesario hacer una prueba en vacío, ya que todo el material filtrante es inalterable al calor como consecuencia del tratamiento previo.
1.5 Cálculos.
El índice de materias celulósicas viene dado por la expresión:

siendo:
C = índice de materias celulósicas en porcentaje de materia seca.
a = peso, en g, del crisol y del residuo después del ataque ácido y secado a 130 °C.
b = peso, en g, del crisol después de la incineración del residuo.
E = peso, en g, de la muestra.
H = humedad de la muestra en porcentaje.
1.6 Observaciones.
1.6.1 En el caso de los cereales y productos derivados, no es necesario extraer la grasa antes del ataque ácido.
1.6.2 Los materiales filtrantes (crisoles filtrantes, arena de mar, placas de porcelana, polvo de tierra cocida) pueden ser utilizados nuevamente. Se separa por tamizado la arena de mar del polvo de tierra cocida; no es necesario limpiar de nuevo con el ácido clorhídrico los crisoles filtrantes, la arena de mar, el polvo de tierra cocida y las placas filtrantes de porcelana.
1.6.3 El tiempo necesario para la determinación del índice de materias celulósicas mediante el empleo de crisoles filtrantes de cuarzo, es aproximadamente de cinco horas, y mediante los crisoles filtrantes de vidrio alrededor de ocho horas.
1.7 Referencia.
1. International Association for Cereal Chemistry (I. C. C.). Standard Nr 113.
2. HUMEDAD
2.1 Principio.
El contenido en agua de un producto se define convencionalmente como la pérdida de masa que experimenta en condiciones determinadas.
El producto se seca a 130 °C bajo presión atmosférica normal, durante una hora y media.
Este método de desecación a 130 °C se aplica a los granos, harinas y otros productos derivados de los cereales, reducidos a partículas de dimensiones inferiores o iguales a 1.700 μ, de las cuales, menos del 10 por 100 serán superiores a 1.000 µ y más del 50 por 100 inferiores a 500 μ.
2.2 Material y aparatos.
2.2.1 Balanza con precisión de 1 mg.
2.2.2 Aparato triturados que no provoque calentamiento, fácil de limpiar y que proporcione partículas de dimensiones especificadas en 2.1.
2.2.3 Pesafiltro metálico o de vidrio con tapadera, y con una superficie útil que permita un reparto de la muestra de 0,3 gramos/cm3, como máximo.
2.2.4 Estufa isoterma de calefacción eléctrica, regulada de tal manera que la temperatura del aire en su interior sea de 130 °C, y que tenga aireación suficiente. La estufa tendrá una capacidad calorífica tal que, regulada previamente a la temperatura de 130 °C, pueda alcanzar de nuevo esa temperatura en menos de media hora, después de colocar simultáneamente en su interior al número máximo de muestras a desecar.
La eficacia de la ventilación se determinará con la ayuda de sémola como material de ensayo que tenga 1 mm como máximo de partícula. La ventilación será tal que secando simultáneamente a 130 °C todas las muestras que la estufa pueda contener, primero durante dos horas y después durante tres horas, los resultados presenten entre ellos una diferencia inferior a 0,15 por 100.
2.2.5 Desecador provisto de placa de porcelana o metálica perforada, conteniendo un agente deshidratante como anhídrido fosfórico, cloruro de calcio o silicagel coloreado en azul.
2.3 Procedimiento.
Introducir 5 g de la muestra en el pesafiltros, tarado después de permanencia en la estufa y de enfriamiento en el desecador. Cerrar el pesafiltros y pesar con aproximación de 1 mg Debe operarse rápidamente.
Tener en la estufa durante hora y media el pesafiltros destapado con la muestra. Transcurrido este tiempo, y operando rápidamente, retirar el pesafiltros de la estufa una vez tapado v colocarlo en el desecador. Pesar en cuanto se enfríe en el desecador.
2.4 Cálculo.
2.4.1 El contenido en agua de la muestra, en porcentaje, es:

en la que:
M = masa inicial, en g, de la muestra.
m = masa, en g, del producto seco.
La media de dos resultados, con una aproximación de 0,05 g por 100, representará la humedad de la muestra.
2.4.2 Dispersión de los resultados. La diferencia resultante entre determinaciones duplicadas de la misma muestra no deberá ser mayor que 0,1 por 100 en valor absoluto. En caso contrario, se repetirá la determinación por duplicado.
2.5 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 34.400 h 5.
2. Métodos de la Asociación Internacional de Química Cerealista (I. C. C.).
3. CENIZAS
3.1 Principio.
3.1.1 Definición. El contenido en cenizas de un producto es el residuo resultante después de su incineración en condiciones determinadas.
Este método es aplicable a los granos, harinas y otros productos derivados de los cereales.
3.2 Material y aparatos.
3.2.1 Balanza analítica con precisión de 0,1 mg.
3.2.2 Horno de mufla eléctrico, con circulación de aire suficiente, con mecanismo de regulación y control de temperatura.
3.2.3 Cápsulas de incineración redondas de fondo plano, preferiblemente de aleación de oro y platino, o bien de cuarzo o de porcelana. El diámetro de las cápsulas será de unos 5 cm. y la altura máxima de 2 cm.
3.2.4 Desecador provisto de llave, con placa perforada de aluminio, conteniendo un agente deshidratante como cloruro de calcio, anhídrido fosfórico o silicagel coloreado en azul.
3.3 Procedimiento.
Pesar 5 g de muestra con aproximación de 10 mg; las restantes pesadas deben hacerse con una aproximación de 0,1 mg.
Inmediatamente antes de usar las cápsulas de incineración, calentarlas en el horno a la temperatura de 910 °C durante 15 minutos. Enfriarlas en el desecador y pesarlas en cuanto alcancen la temperatura ambiente.
Introducir la muestra pesada en la cápsula repartiéndola en una capa de espesor uniforme, sin comprimirla. Colocar la cápsula a la entrada del horno con la puerta abierta, y dejar que arda. Cuando las llamas se extingan, empujar la cápsula al interior del horno y cerrar la puerta del mismo. Una vez cerrada la puerta del horno debe mantenerse en él una corriente de aire suficiente, que no sea tan fuerte como para arrastrar la sustancia fuera de las cápsulas.
La incineración se continúa hasta lograr la combustión total de la muestra, incluso de las partículas carbonosas que pueden quedar incrustadas en las cenizas. Dar por terminada la incineración cuando el residuo es prácticamente blanco o gris después del enfriamiento. Sacar las cápsulas del horno y dejarlas enfriar en el desecador. Pesarlas tan pronto alcancen la temperatura ambiente
La temperatura de incineración es de 910 °C.
3.4 Cálculo.
3.4.1 El porcentaje de cenizas sobre materia natural se obtiene por la fórmula siguiente:

En la que:
P = peso en g de la cápsula con la muestra.
P1= peso en g de la cápsula con las cenizas.
P2 = peso en g de la cápsula vacía.
3.4.2 El porcentaje de cenizas sobre materia seca se obtiene relacionando el valor de contenido en cenizas obtenido sobre materia natural con el valor de contenido en humedad, según la fórmula siguiente:

3.4.3 Límite de errores. Cuando el contenido de cenizas no rebase el 1 por 100 de la muestra, la diferencia de los resultados de un ensayo efectuado por duplicado no deberá ser superior al 0,02. Si el contenido de cenizas rebasa el 1 por 100, la diferencia no deberá ser superior al 2 por 100 de dicho contenido Si es superior, se repetirá la determinación.
3.4.4 Expresión de los resultados. El contenido de cenizas se expresa por 100 partes de sustancia seca y con dos cifras decimales.
3.5 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 34.400 h 8.
2. Métodos de la Asociación Internacional de Química Cerealista (I. C. C.).
4. PROTEÍNA
4.1 Principio.
El contenido en proteína bruta de un producto es el resultado de multiplicar el contenido en nitrógeno, determinado por el procedimiento Kjeldahl por un factor de transformación del nitrógeno en proteína.
Este método es aplicable a los granos, harinas y otros derivados de los cereales.
4.2 Material y aparatos.
4.2.1 Matraces Kjeldahl de 500 a 800 ml.
4.2.2 Batería de ataque
4.2.3 Batería de destilación o aparato de destilación.
4.3 Reactivos.
4.3.1 Ácido sulfúrico concentrado.
4.3.2 Sulfato potásico anhidro.
4.3.3 Sulfato de cobre.
4.3.4 Disolución de hidróxido sódico al 30 por 100 (p/v) en agua.
4.3.5 Disolución de ácido sulfúrico 0,1N.
4.3.6 Disolución de hidróxido sódico 0,1N.
4.3.7 Disolución de indicador: disolver 0,3 g de rojo de metilo en 100 ml de alcohol etílico del 95 por 100.
4.4 Procedimiento.
Pesar un g de muestra, molida de forma que las partículas sean inferiores a 500 μ, e introducirla en un matraz Kjeldahl. Añadir 10 g de sulfato potásico y 0,1 g de sulfato de cobre. Agregar 20 ml de ácido sulfúrico concentrado y mezclar todo hasta que toda la sustancia esté mojada por el ácido. Iniciar el ataque a fuego lento, para evitar que la espuma arrastre el producto al cuello del matraz. Cuando desaparezca la espuma, hacer hervir vigorosamente hasta que la disolución quede limpia y prolongar todavía el ataque otros 30 minutos.
Dejar enfriar. Añadir unos 200 ml de agua. Agregar 80 ml de hidróxido sódico al 33 por 100 y proceder al destilado. El líquido que destila se recoge en un vaso que contenga 20 ml de SO4H2 N/10 y una gota de disolución de indicador, añadiéndose nuevamente una cantidad conocida de ácido sulfúrico N/10 si virase de color durante la destilación. La cantidad de destilado a recoger es de unos 150 ml, dándose por acabada la destilación cuando el líquido que se destila no haga virar a azul el papel rojo de tornasol.
Acabada la destilación, valorar el exceso de ácido sulfúrico con disolución valorada de hidróxido sódico 0,1N.
Efectuar una prueba en blanco de destilación y valoración para controlar la pureza de los reactivos.
4.5 Cálculo.
4.5.1 El porcentaje de proteína bruta sobre sustancia natural es:

En la que:
V = volumen en ml de disolución de SO4H2 N/10 empleado para recoger el nitrógeno amoniacal destilado.
f = factor de la disolución de SO4H2 N/10.
V1 = volumen en ml de disolución de NaOH N/10 necesario para neutralizar el ácido sulfúrico existente al final de la destilación.
f1 = factor de la disolución de NaOH N/10.
F = factor de transformación de nitrógeno en proteína. Para el trigo y derivados es de 5,7 y para los restantes cereales es de 6,25.
P = peso de la muestra.
4.5.2 El porcentaje de proteína bruta sobre sustancia seca se determina teniendo en cuenta el contenido en humedad.
4.5.3 Dispersión de los resultados. Se considerarán concordantes las determinaciones duplicadas cuando los resultados expresados en porcentaje difieran en menos de 0,25.
4.6 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 34.400 h 7.
2. Métodos de la Asociación Internacional de Química Cerealista (I. C. C.).
5. GRASA
5.1 Principio.
El contenido en grasa bruta de un producto se define convencionalmente como la parte del mismo extraíble por éter etílico en condiciones determinadas. Incluye, además de la grasa, otras muchas sustancias solubles en éter etílico, como son: ceras, pigmentos, vitaminas, etc.
Este método es aplicable a los granos, harinas y otros productos derivados de los cereales.
5.2 Material y aparatos.
5.2.1 Extractor tipo Soxhlet.
5.2.2 Balanza analítica con precisión de 0,1 mg.
5.2.3 Estufa de desecación, graduada a 100 °C.
5.2.4 Desecador con placa de porcelana o metálica perforada, conteniendo un agente deshidratante, como anhídrido fosfórico o silicagel.
5.2.5 Cartuchos de extracción.
5.2.6 Matraces de 100 a 150 ml, adaptables al extractor.
5.2.7 Batería de extracción, baño de agua.
5.3 Reactivos.
Éter etílico.
5.4 Procedimiento.
Pesar de 5 a 10 g de muestra, molida de forma que pase por un tamiz de 500 µ y desecada a 100 °C, e introducirlos en un cartucho que se tapona con algodón. Tarar el matraz, desecado en la estufa y enfriado en el desecador. Introducir el cartucho en el extractor, añadir éter etílico una vez conectado el matraz y proceder a la extracción, continuándola hasta que el éter sea incoloro; son suficientes 4 horas a una velocidad de destilación de 4 a 5 gotas/s, y 16 horas para 2 a 3 gotas/s.
Sacar el cartucho del extractor y recuperar el éter. Llevar el matraz con el extracto y el resto del disolvente a la estufa de desecación a 100 °C y tenerlo media hora. Dejar enfriar el matraz en el desecador y, en cuanto alcance la temperatura ambiente, pesarlo.
5.5 Cálculo.
El porcentaje de grasa bruta sobre sustancia seca viene dado por la fórmula:

En la que:
P1 = peso, en g, del matraz con el extracto etéreo.
P2 = peso, en g, del matraz vacío.
P = peso, en g, de la muestra empleada.
5.6 Referencia.
1. American Association of Cereal Chemists. Cereal Laboratory Methods, 1967. Método 30-20.
6. ÍNDICE DE MALTOSA
6.1 Principio.
El índice de maltosa es una medida relacionada con la capacidad de producción de gas de un trigo.
6.2 Material y aparatos.
6.2.1 Matraz erlenmeyer con tapón de 250 ml.
6.2.2 Baño de agua.
6.2.3 Bureta graduada de 50 ml de capacidad.
6.2.4 Matraz erlenmeyer de 100 ml.
6.2.5 Mechero Bunsen y trípode.
6.3 Reactivos.
6.3.1 Ácido sulfúrico al 20 por 100.
6.3.2 Tungstato sódico al 15 por 100.
6.3.3 Solución de sulfato de cobre (69,28 g/l).
6.3.4 Solución de 350 g de sal de Seignette (tartrato sódico potásico) y 100 g de sosa cáustica en 1 litro de agua.
6.3.5 Indicador: Solución acuosa de azul de metileno al 1 por 100.
6.4 Procedimiento.
Pesar 15 g de harina y colocarlos en un matraz erlenmeyer con tapón. Añadir 95 ml de agua destilada e introducir el matraz en un baño de agua manteniendo a 27 °C de temperatura. Dejar en el baño durante una hora, agitar bien a intervalos de 15 minutos. Sacar el matraz del baño y añadir 15 ml de ácido sulfúrico al 20 por 100 y 3,5 ml de tungstato sódico al 15 por 100. Filtrar. Prescindir del residuo y colocar el líquido en una bureta graduada de 50 ml de capacidad. Introducir en un matraz erlenmeyer 5 ml de solución de sulfato de cobre (69,28 g/l) y 6 ml de una solución de 350 g de sal de Seignette (tartrato sódico potásico) y 100 g de sosa cáustica en un 1 litro de agua Colocar el matraz en un trípode sobre un mechero Bunsen y verter en él desde la bureta 20 ml del líquido filtrado. Cuando hierva, añadir 5 gotas de indicador (solución acuosa de azul de metileno al 1 por 100). Verter gradualmente líquido desde la bureta hasta que la solución del matraz ha perdido por completo la coloración azul. Observar en la probeta la cantidad de líquido vertido y buscar en la tabla 6.1 el índice de maltosa de la harina.
6.5 Referencia.
1. Análisis de Cereales y Derivados. Ministerio de Agricultura, 1957, páginas 56-57.
TABLA 6.1
Índice de maltosa
| Líquido vertido | Índice de maltosa | Líquido vertido | Índice de maltosa |
|---|---|---|---|
| 20,0 | 2,69 | 35,5 | 1,49 |
| 20,5 | 2,63 | 36,0 | 1,47 |
| 21,0 | 2,56 | 36,5 | 1,46 |
| 21,5 | 2,50 | 37,0 | 1,45 |
| 22,0 | 2,44 | 37,5 | 1,42 |
| 22,5 | 2,38 | 38,0 | 1,40 |
| 23,0 | 2,33 | 38,5 | 1,38 |
| 23,5 | 2,28 | 39,0 | 1,36 |
| 24,0 | 2,23 | 39,5 | 1,34 |
| 24,5 | 2,18 | 40,0 | 1,32 |
| 25,0 | 2,14 | 40,5 | 1,30 |
| 25,5 | 2,10 | 41,0 | 1,29 |
| 26,0 | 2,06 | 41,5 | 1,27 |
| 26,5 | 2,02 | 42,0 | 1,26 |
| 27,0 | 1,99 | 42,5 | 1,24 |
| 27,5 | 1,95 | 43,0 | 1,23 |
| 28,0 | 1,91 | 43,5 | 1,21 |
| 28,5 | 1,87 | 44,0 | 1,20 |
| 29,0 | 1,84 | 44,5 | 1,18 |
| 29,5 | 1,80 | 45,0 | 1,17 |
| 30,0 | 1,77 | 45,5 | 1,16 |
| 30,5 | 1,74 | 46,0 | 1,15 |
| 31,0 | 1,72 | 46,5 | 1,13 |
| 31,5 | 1,69 | 47,0 | 1,12 |
| 32,0 | 1,66 | 47,5 | 1,11 |
| 32,5 | 1,63 | 48,0 | 1,10 |
| 33,0 | 1,61 | 48,5 | 1,09 |
| 33,5 | 1,58 | 49,0 | 1,08 |
| 34,0 | 1,56 | 49,5 | 1,06 |
| 34,5 | 1,54 | 50,0 | 1,05 |
| 35,0 | 1,52 |
7. ACIDEZ GRASA
7.1 Principio.
Neutralización de los ácidos grasos libres con hidróxido sódico. Se mide la rancidez hidrolítica que se utiliza como índice de deterioro en almacenamiento.
Aplicable a granos de cereales y harinas.
7.2 Material y aparatos.
7.2.1 Extractor tipo Soxhlet.
7.2.2 Balanza analítica con precisión de 0,1 mg.
7.2.3 Cartuchos de extracción.
7.2.4 Matraces de 100 a 150 ml adaptables al extractor.
7.2.5 Batería de extracción con baño de agua.
7.2.6 Pipetas de 50 ml.
7.2.7 Bureta de 50 ml.
7.3 Reactivos.
7.3.1 Éter de petróleo p.e. 35-60 °C.
7.3.2 Benceno-alcohol-fenolftaleína (BAF). Mezclar partes iguales en volumen de benceno y de alcohol etílico del 95 por 100. Añadir 0,2 g de fenolftaleína por litro para obtener una solución al 0,02 por 100.
7.3.3 Hidróxido potásico. Preparar una solución 0,0178N (1 ml = 1 mg KOH) con hidróxido potásico libre de CO2.
7.3.4 Patrón de color.
7.3.4.1 Colocar 50 ml de agua en un matraz del mismo tipo en que se va a hacer la valoración. Añadir gota a gota una solución de dicromato potásico al 0,05 por 100 hasta que tome la coloración de la solución que se va a valorar. Añadir 2,5 mililitros de una solución recién preparada de permanganato potásico al 0,01 por 100 y se mezcla. El color final de la valoración debe ser semejante a éste.
7.3.4.2 El color patrón para la valoración de la prueba en blanco se obtiene añadiendo 2,5 ml de permanganato al 0,01 por 100 a 50 ml de agua.
7.4 Procedimiento.
Para que los resultados sean más precisos, el contenido en humedad de los granos no debe exceder del 11 por 100. Se ha comprobado que mayores contenidos en humedad en el momento de la extracción aumentan significativamente los valores de acidez grasa.
Moler por los menos 40 g de una muestra representativa para granos pequeños, tales como el trigo, o 200 g para granos más grandes, tales como el maíz. Preferiblemente, moler la muestra de tal forma que el 90 por 100 o más pase a través del tamiz de 500 μ. Una vez molida la muestra se somete a extracción antes de 1 hora para evitar cambios causados por enzimas lipolíticos. Extraer 10 g de muestra sólida, como en 5.4, utilizando éter de petróleo, Evaporar el éter de petróleo del extracto y redisolver el extracto en el matraz de extracción con 50 ml de solución de BAF. Valorar la solución extraída con hidróxido potásico 0,0178N hasta alcanzar el punto de color patrón.
7.4.1 Hacer la prueba en blanco valorando 50 ml de solución BAF, hasta alcanzar el punto de color patrón 7.3.4.2.
7.5 Cálculo.
Expresar la acidez grasa como los mg de KOH requeridos para neutralizar los ácidos grasos libres de 100 g de grano sobre sustancia seca por la fórmula

donde:
V = volumen en ml de KOH 0,0178N utilizado para valorar la muestra extraída.
V1 = volumen en ml de KOH 0,0178N utilizado para la valoración en blanco.
H = peso en g de agua en 100 g de muestra.
7.6 Observaciones.
En caso de granos con altos valores de acidez grasa se forman a veces emulsiones durante la valoración que enmascaran parcialmente el punto de color final. Cuando aparecen emulsiones, añadir 50 ml adicionales de solución BAF para asegurar una solución clara. En este caso el valor de la prueba en blanco es el doble del valor determinado sobre 50 ml.
7.7 Referencia.
1. American Association of Cereal Chemistry Cereal Laboratory Methods, 1967. Método 02-01.
8. AGENTES OXIDANTES
(Reacción con yoduro potásico)
8.1 Principio.
Este método detecta todos los agentes oxidantes que se adicionan generalmente a la harina, para mejorar sus propiedades de panificación, excepto percloratos y peróxido de benzoilo.
8.2 Material y aparatos.
8.2.1 Matraz erlenmeyer de 500 ml.
8.2.2 Centrífuga.
8.3 Reactivos.
8.3.1 Yoduro potásico 10 por 100.
8.3.2 Ácido sulfúrico (1 ± 10) v/v.
8.4 Procedimiento.
Colocar 50 g de muestra en un matraz erlenmeyer de 500 mililitros, añadir 200 ml de agua a la temperatura ambiente, agitar bien y dejar reposar 1 hora, aproximadamente, con agitados frecuentes. Filtrar o centrifugar. A 5 ml del filtrado añadir 5 mililitros de solución de KI y 5 ml de H2SO4 (1 + 10) v/v. Soluciones amarillas o pardas indican la presencia de agentes oxidantes.
8.5 Referencia.
1. American Association of Cereal Chemistry. Cereal Laboratory Methods. 1967. Método 48-02.
9. BROMATOS Y IODATOS EN LA HARINA
(Método cualitativo)
9.1 Principio.
Este método sirve para determinar la presencia de bromato y iodato en la harina, que actúan como mejorantes.
9.2 Material y aparatos.
9.2.1 Placa de Petri de un área aproximada de 100 cm2.
9.2.2 Tamiz número 60.
9.3 Reactivos.
9.3.1 Para el bromato y iodato solución de HCl en agua (1 + 7) v/v y KI (1 por 100) mezclados a volúmenes iguales.
9.3.2 Para iodato solución de 1 volumen de KSCN (1 por 100) y 4 volúmenes de solución de HC1 en agua (1 + 32) v/v mezclados.
9.4 Procedimiento.
9.4.1 Bromatos y iodatos.
Cubrir el fondo del recipiente con el reactivo KI-HCl. Cerner uniformemente con el tamiz número 60 sobre el reactivo, aproximadamente 4 g de harina a ensayar. Alternativamente, cerner harina sobre la superficie del recipiente seco y esparcir la mezcla de reactivo sobre la harina con un frasco pulverizador hasta que todas las partículas estén humedecidas. La aparición de manchas negras o purpúreas después de la adición del reactivo indica la presencia de bromato o iodato.
9.4.2 lodatos.
9.4.2.1 (Aplicable a 10 p.p.m. o más). Distribuir suavemente, aproximadamente, g de harina sobre el fondo de una placa de Petri y cubrir completamente con el reactivo KSCN-HC1 recientemente preparado.
9.4.2.2 (Aplicable a 1-10 p.p.m.) Proceder como para bromatos y iodatos, pero usar el reactivo KSCN-HC1.
9.5 Referencia.
1. Association of Official Agricultural Chemists. Official Methods of Analysis. 1970 14.040 y 14.041, pág. 218.
10. PERÓXIDO DE BENZOILO
(Método cualitativo con bencidina)
10.1 Principio.
El peróxido de benzoilo da con solución etanólica de bencidina, coloración pardo-verdosa.
10.2 Material y aparatos.
10.2.1 Placa de vidrio
10.2.2 Pulverizador.
10.3 Reactivos.
Disolver 1,5 g de bencidina (base libre) en 50 ml de etanol 96 por 100. Calentar la solución a 50-60° en un baño de agua antes de usarla.
10.4 Procedimiento.
Verter el reactivo sobre una capa de harina en una placa de vidrio. Si aparecen manchas pardo-verdosas indica que la harina está tratada con peróxido de benzoilo. Observar mejor las manchas por detrás del vidrio y, en general, verter el reactivo pulverizando.
10.5 Referencia.
1. American Association of Cereal Chemists. Cereal Laboratory Methods, 1967. Método 48-05.
11. ÍNDICE DE PELSHENKE
11.1 Principio.
El índice de Pelshenke proporciona una valoración indirecta de la calidad panadera de los trigos, estando relacionado tanto con la capacidad de producción de gas como con la capacidad de retención del mismo.
11.2 Material y aparatos.
11.2.1 Bario de agua.
11.2.2 Vasos de forma baja de 150 ml.
11.3 Reactivos.
11.3.1 Suspensión de levadura panaria. Formar una papilla espesa con 10 g de levadura y agua destilada, añadiendo más agua hasta completar 100 ml.
11.4 Procedimiento.
Pesar 10 g de trigo molido que pase por el tamiz de 1 mm y colocarlo en una cápsula de porcelana. Añadir 5,5 ml de la suspensión de levadura y amasar con una espátula. Dividir la masa en dos partes aproximadamente iguales y darle forma de bola compacta entre las palmas de las manos.
Introducir las bolas así formadas en sendos vasos de forma baja de 150 ml, que contengan 75 ml de agua a 32 °C. Colocar los vasos en un baño de agua a dicha temperatura. Por la acción de los gases producidos en la fermentación, las bolas suben a la superficie, en la cual permanecen un tiempo variable, hasta que se rompen y caen pedazos al fondo del vaso.
11.5 Cálculo.
Medir el tiempo en minutos transcurrido desde el momento de introducir la bola en el vaso hasta que se produce la disgregación. La media de las dos determinaciones constituye el «índice de Pelshenke».
11.6 Referencias.
1. Análisis de Cereales y Derivados. Ministerio de Agricultura, 1957, pág. 35.
2. American Association of Cereal Chemists Cereal Laboratory Methods, 1967. Método 56-50.
12. GLUTEN
12.1 Principio.
Complejo de proteínas insolubles en agua que forman, por arrastre del almidón de la harina mediante lavado, una masa gomosa muy extensible.
Este método se aplica para la determinación del contenido en gluten de la harina de trigo y sémolas.
12.2 Material y aparatos.
12.2.1 Balanza con precisión de 0,01 g.
12.2.2 Extractor de gluten con disco excéntrico y mecanismo tensor para gasa de seda; velocidad del disco excéntrico: 80 r.p.m.
12.2.3 Recipiente para agua con gasto regulable.
12.2.4 Cronómetro.
12.2.5 Tamiz de madera, de 30 por 40 cm, con gasa para sémola número 56.
12.2.6 Placa de vidrio esmerilado 40 x 40 cm.
12.2.7 Guantes de caucho delgado y de superficie lisa.
12.2.8 Prensa para gluten, sistema Berliner, con distancia entre placas de 2,4 mm.
12.2.9 Cápsula de porcelana barnizada interiormente o de metal esmaltado, de 10 a 15 cm de diámetro.
12.2.10 Espátula de 18 a 20 cm de longitud.
12.3 Reactivos.
12.3.1 Disolución al 2 por 100 de cloruro de sodio (pH 6,2). Disolver 200 g de cloruro de sodio, de calidad reactivo para análisis, en 10 litros de agua destilada. Añadir 7,54 g de KR2PO4 y 1,40 g de Na2HPO4 × 2H2O, de calidad reactivo para análisis. La disolución se preparará cada día que se utilice.
12.3.2 Solución de yodo, aproximadamente N/1000; sirve para comprobar la presencia de almidón.
12.4 Procedimiento.
Pesar 10 g de harina con una aproximación de ± 0,01 g y colocarla en una cápsula de porcelana. Añadir gota a gota 5,5 ml de disolución de cloruro sódico removiendo continuamente la harina con la espátula. Después de haber añadido a la harina toda la disolución de cloruro de sodio, comprimir la mezcla cuidando de no perder nada de harina. La masa adherida a la pared de la cápsula se añade a la bola de masa.
Homogeneizar la masa enrollándola con la palma de la mano sobre la placa de vidrio esmerilado hasta que tenga una longitud de 7 a 8 cm, volviéndola a dar entonces la forma de bola y se repite el amasado en la misma forma hasta un total de cinco veces. La mano que efectúa la homogeneización estará revestida de un guante de caucho que proteja la masa del calor y de la transpiración de la mano.
Colocar la bola de masa sobre la gasa de seda ligeramente tensa del extractor de gluten. Mojar la masa con unas gotas de solución de cloruro de sodio, colocando luego en su sitio el disco excéntrico. Lavar durante 10 minutos, debiéndose gastar unos 400 ml de solución de cloruro de sodio.
Cuando no se disponga del aparato extractor de gluten, se sustituirá el anterior paso por un lavado a mano. Para ello, dejar caer gota a gota la solución de cloruro de sodio, que debe tener una temperatura de 18 °C, sobre la palma de la mano. El ritmo de goteo debe ser tal que aproximadamente 0,75 litros de la disolución desagüe en 8 minutos. Durante este tiempo arrollar y prensar alternativamente la masa y estirarla siete veces de forma que se parta en dos trozos que se juntan en seguida. La duración del lavado depende del contenido de la masa en gluten; sin embargo, debe ser aproximadamente la misma siempre y no rebasar los 8 minutos.
Al lavado mecánico del gluten sigue un lavado a mano cuya duración, en general, no debe exceder de 2 minutos. Se puede considerar terminada la extracción de gluten tan pronto como al amasar la bola de gluten con la disolución fresca de cloruro de sodio no se encuentren más que trazas de almidón en el agua escurrida. Para comprobar la presencia de almidón en el líquido de lavado, utilizar una disolución de yodo 0,001 N.
Desprender de la bola de gluten la mayor parte de la disolución de lavado adherente cogiendo el gluten con la punta de los dedos de una mano y sacudiéndolo tres veces brevemente, pero con fuerza. Estirar a continuación, suavemente, el gluten en lámina delgada, manteniéndolo entre los dedos, y llevarlo a la prensa, cerrando ésta. Abrirla a los 5 segundos y pasar la lámina de gluten a posición seca sin deformarla. Prensarla otra vez. Hacer esta operación quince veces, secando bien las superficies de vidrio después de cada prensada.
Pesar el gluten en la balanza con aproximación de 0,01 g.
12.5 Cálculo.
12.5.1 Gluten húmedo. El peso obtenido multiplicado por diez da el porcentaje de gluten húmedo. Las determinaciones duplicadas se consideran concordantes cuando no difieran en más de 0,5 por 100 de contenido en gluten. Si la desviación es mayor, hacer una tercera determinación y tomar la medida de las tres efectuadas como expresión del contenido en gluten. Si la desviación hallada entre los valores más alto y más bajo en los tres ensayos es mayor del 1 por 100, proceder a hacer una cuarta determinación.
12.5.2 Gluten seco. La bola de gluten húmedo obtenida en la determinación anterior se deseca en la estufa a temperatura de 100 °C hasta peso constante. Dejarla enfriar y pesar.
El peso obtenido multiplicado por diez da el porcentaje de gluten contenido en la harina.
12.6 Referencias.
1. Instituto de Racionalización del Trabajo. Una Norma Española 34.400 h 6.
2. Métodos de la Asociación Internacional de Química Cerealista (I. C. C.).
13. FARINÓGRAFO BRABENDER
13.1 Principio.
El método se aplica para la determinación de la absorción de agua y el comportamiento al amasado de una harina de trigo. El farinógrafo mide y registra la resistencia de una masa al amasado. Esta resistencia se llama consistencia. La absorción de agua se define como el porcentaje de agua respecto al peso de harina que es necesario añadir para obtener una masa de consistencia determinada.
13.2 Material y aparatos.
13.2.1 Farinógrafo Brabender con tanque de circulación de agua.
Calibración del farinógrafo.
Velocidad de la paleta rápida: 90 ± 3 r.p.m.
Par: 100 ± 2 g cm/U.B.
Velocidad del papel: 1,00 ± 0,03 cm/min.
Bureta graduada de 135 ml a 225 ml.
13.2.2 Balanza de sensibilidad: ± 0,1 g.
13.2.3 Espátula de plástico blanco.
13.3 Reactivos.
13.3.1 Agua destilada.
13.4 Procedimiento.
Determinar el contenido de humedad de la harina como en 2. Hacer circular el agua por el termostato y el farinógrafo al menos 1 hora antes de usar el instrumento. Durante el ensayo, la temperatura del agua y de la amasadora deberá ser de 30° ± 0,2°.
Lubricar la amasadora con una gota de agua entre la pared posterior y cada una de las paletas. Ajustar la posición de los pesos de la balanza paca obtener una deflección cero del indicador con las paletas girando en vacío. Ajustar el brazo de la pluma de tal forma que coincidan las lecturas en el sector de la balanza y en el pape móvil. Ajustar el amortiguador de tal manera que con el motor girando el tiempo requerido por el indicador de la balanza para ir de 1.000 a 100 U.B. sea de 1,0 ± 0,2 s.
Poner en la amasadora el peso equivalente a 300 ± 0,1 g de harina con el 15 por 100 de humedad. Tapar la amasadora. Llenar la bureta con agua a 30° ± 5°.
Si fuese necesario, llevar la harina a 30° ± 10°.
Colocar el papel de tal manera que la pluma esté en contacto con una línea de 9 min. Mezclar durante 1 minuto. Comenzar a añadir agua de la bureta en la esquina delantera de la derecha de la amasadora cuando la pluma cruce la línea de 0 minutos. Añadir agua en cantidad suficiente para que la consistencia máxima de la masa sea de 500 U.B.
Cuando la masa se adhiera a las paredes de la amasadora, rasparla con una espátula de plástico. Cubrir la amasadora hasta el final del ensayo.
Si la cantidad de agua utilizada en el ensayo no se ha añadido en un intervalo de 25 segundos o si la consistencia máxima de la masa difiere de 500 ± 20 U.B., se repite el ensayo corrigiendo la cantidad de agua y añadiéndola en 25 segundos de forma que la masa adquiera una consistencia máxima de 500 ± 20 U.B.
Una vez alcanzada la consistencia máxima, continuar el ensayo durante 12 minutos.
13.5 Cálculo.
13.5.1 Absorción de agua. Calcular la absorción de agua sobre una base del 15 por 100 de humedad como sigue:

V = volumen en ml de agua añadida para obtener una masa con una consistencia máxima de 500 U.B.
P = peso en g de harina utilizada, equivalente a 300 g con el 15 por 100 de humedad.
Determinar la absorción con una aproximación de 0,1 por 100.
13.5.2 Tiempo de desarrollo. Es el período de tiempo comprendido desde el comienzo del amasado hasta el punto de la curva inmediatamente anterior al primer signo de decaimiento. Expresar este tiempo con una aproximación de 0,5 minutos. En el caso, poco frecuente, de que aparezcan dos máximos, emplear el segundo para medir el tiempo de desarrollo.
13.5.3 Grado de decaimiento. Es la diferencia en U.B. entre el centro de la curva en el máximo y el centro de la curva 12 minutos después de este máximo. Expresar el decaimiento con una aproximación de 5 U.B.
13.6 Observaciones.
Se obtienen los siguientes valores para el coeficiente de variación de la determinación simple correspondiente a un solo farinógrafo:
Absorción de agua: 0,55%.
Tiempo de desarrollo de la masa: 8,9%.
Grado de decaimiento: 7,5%.
13.7 Referencia.
1. Métodos de la Asociación Internacional de Química Carealista (I. C. C.).
14. ALVEÓGRAFO CHOPIN
(Provisional)
14.1 Principio.
En el ensayo con el alveógrafo, la masa se extiende bidimensionalmente formando un alveolo, por efecto de la fuerza debida a la presión del aire que se insufla por debajo de una lámina de masa obtenida en condiciones normalizadas. Con este ensayo se imita a gran escala la formación de alveolos en el seno de la masa por el dióxido de carbono producido por las levaduras durante la fermentación.
Las dimensiones y la forma de las curvas obtenidas y el volumen del alveolo en e! momento de la rotura son una guía de las características de panificación de la harina.
14.2 Material y aparatos
14.2.1 Alveógrafo de Chopin (con depósito de circulación de agua).
Calibración del alveógrafo:
Velocidad de la paleta amasadora: 60 ± 3 r.p.m.
Altura de las guías: 1,2 ± 0,1 cm.
Diámetros del rodillo: 3,3 ± 0,07 cm; 4,0 ± 0,10 cm.
Diámetro del cortador: 4,55 ± 0,05 cm.
Diámetro de la pieza de masa antes del ensayo: 5,50 ± 0,05 cm.
Volumen de la pera de goma: 20 ± 5 ml.
Volumen de la bureta: 625 ± 3 ml.
Flujo de agua en la bureta: 23,0 ± 0,5 s.
Velocidad del papel, 0,30 cm: 55 ± 1 s ó 0,55 + 0,01 cm/s.
14.2.2 Bureta de 200 ml de capacidad.
14.2.3 Balanza de sensibilidad ± 0,1 g.
14.2.4 Matraz erlenmeyer 250 ml.
14.2.5 Reloj avisador.
14.2.6 Planímetro.
14.3 Reactivos.
14.3.1 Solución de cloruro sódico. Disolver 25 g de cloruro sódico reactivo análisis, en agua destilada y llevar hasta 1 litro.
14.3.2 Aceite de cacahuete.
14.4 Procedimiento.
Determinar el contenido de humedad de la harina como en 2.3.
Si fuese necesario, llevar la temperatura de la harina a 20° ± 5°. Poner el termostato en funcionamiento con antelación suficiente para asegurar que durante el ensayo la temperatura de la amasadora y del alveógrafo sea de 25° ± 0,2°. Antes y durante el ensayo, comprobar las temperaturas.
Verter de la bureta al matraz erlenmeyer el volumen de solución de cloruro sódico equivalente a 50 ml. por cada 1.000 g de harina con el 15,0 por 100 de humedad. Este volumen puede encontrarse en la tabla 14.1.
Poner en la amasadora 250 ± 0,1 g de harina y colocar el suplemento de la amasadora en su posición. Poner en marcha el motor de la amasadora en su posición de marcha adelante, poner en marcha el reloj y añadir a la harina la solución de cloruro sódico vertiéndola sobre el eje de la paleta amasadora. Esta adición deberá hacerse en 15 segundos. Esperar a que se forme la masa. Después de 60 segundos retirar el suplemento de la amasadora y colocar la tapa sobre la amasadora. Después de 6 minutos para el motor de la amasadora.
Poner en marcha el motor en su posición de marcha atrás. Abrir la ranura de extrusión y colocar unas pocas gotas de aceite sobre la placa receptora. Desechar los 2 cm primeros de masa.
Cuando la lámina de masa alcance las muescas de la placa receptora, cortar con dos rápidos cortes frente a las guías. Deslizar las piezas de masa sobre la placa de vidrio previamente aceitado.
Repetir la operación anterior tres veces más y dejar una quinta pieza de masa sobre la placa receptora. Parar el motor de la amasadora.
Colocar el rodillo, cuya superficie está aceitada, entre las dos piezas de masa y moverle a lo largo de los raíles 12 veces, primero lentamente y luego rápidamente. Repetirlo con la tercera y cuarta pieza. Cortar cada pieza con el cortador circular y colocarlas ordenadamente sobre las bandejas aceitadas en la cámara del alveógrafo. Pasar el rodillo y cortar la quinta pieza en la misma forma.
Colocar el papel sobre el tambor registrador. Llenar la pluma, trazar la línea de cero y volver el tambor a la posición inicial. Comprobar que el nivel de agua en la bureta está en el cero. Comprobar que la manilla está en la posición 1. Aceitar el obturador y el plano de la base de la prensa 26 minutos después del comienzo del amasado, desenroscar el cuello de la prensa dos revoluciones Retirar el collar pequeño y el obturador. Con la espátula deslizar cuidadosamente la primera pieza de masa sobre el centro de la base de la prensa. Volver a colocar el obturador y el collar Girar el collar grande dos revoluciones en 20 segundos. Esperar 5 segundos. Retirar el collar pequeño y el obturador. Girar la manilla a la posición 2. Elevar la vasija de agua. Girar la válvula de aire a su posición horizontal. Comprimir la pera de goma. Girar la válvula de aire a su posición vertical. Soltar la pera de goma. Girar la manilla a la posición 3, comenzando la formación del alveolo y la rotación del tambor registrador.
Cuando el alveolo se rompa, girar inmediatamente la manilla a la posición 4. Anotar el nivel de agua en la bureta. Bajar la vasija de agua. Girar la manilla a la posición 1 y volver a colocar la pluma. Desenroscar el collar grande dos revoluciones y retirar la masa. Repetir el ensayo con las cuatro piezas de masa restantes. Si un alvéolo o la curva es claramente anormal debe desecharse la curva.
14.5 Cálculo.
14.5.1 Altura de la curva H. Es la media de las alturas máximas en milímetros de las cinco curvas. El valor P, tenacidad de la masa, puede calcularse multiplicando H por 1,1.
14.5.2 Longitud de la curva L. Es la longitud media, en milímetros, de las cinco curvas. Se miden a lo largo de la línea de cero, desde el comienzo de la curva hasta el punto correspondiente a la vertical trazada por el punto de la curva donde la presión desciende más bruscamente debido a la ruptura del alveolo.
14.5.3 Área de la curva S. Es el área media en cm2 de las curvas. Para determinar esta área, trazar una curva media de las cinco. Si las curvas son diferentes medir la altura de la curva en el máximo, en el medio y hacia el final de cada curva. Marcar los valores medios sobre el gráfico en los puntos apropiados y trazar la curva media también sobre el gráfico conservando la forma característica de la curva.
14.5.4 Índice de inflamiento G. Es el valor medio de las lecturas de la bureta tomadas cuando el alveolo se rompe, que equivalen a la raíz cuadrada del volumen de aire usado para inflar el alveolo.
14.5.5 Trabajo de deformación W. Es el trabajo mecánico, en ergios, usado para inflar el alveolo. Se calcula por la fórmula siguiente:

14.6 Referencia.
1. Grupo de Estudio. Ensayo físico de la masa. Asociación Internacional de Química Cerealista (I. C. C.).
TABLA 14.1
Volumen de la solución de cloruro sódico
|
Humedad ‒ Porcentaje |
Volumen ‒ ml |
Humedad – Porcentaje |
Volumen _ ml |
|---|---|---|---|
| 8,0 | 156,1 | 11,8 | 139,2 |
| 8,2 | 155,2 | 12,0 | 138,3 |
| 8,4 | 154,4 | 12,2 | 137,5 |
| 8,6 | 153,5 | 12,4 | 136,6 |
| 8,8 | 152,6 | 12,6 | 135,7 |
| 9,0 | 151,7 | 12,8 | 134,8 |
| 9,2 | 150,8 | 13,0 | 133,9 |
| 9,4 | 149,9 | 13,2 | 133,0 |
| 9,6 | 149,0 | 13,4 | 132,1 |
| 9,8 | 148,1 | 13,6 | 131,2 |
| 10,0 | 147,2 | 13,8 | 130,3 |
| 10,2 | 146,3 | 14,0 | 129,4 |
| 10,4 | 145,5 | 14,2 | 128,6 |
| 10,6 | 144,6 | 14,4 | 127,7 |
| 10,8 | 143,7 | 14,6 | 126,8 |
| 11,0 | 142,8 | 14,8 | 125,9 |
| 11,2 | 141,9 | 15,0 | 125,0 |
| 11,4 | 141,0 | 15,2 | 124,1 |
| 11,6 | 140,1 | 15,4 | 123,2 |
| 15,6 | 122,3 | 17,8 | 112,5 |
| 15,8 | 121,4 | 18,0 | 111,7 |
| 16,0 | 120,6 | 18,2 | 110,8 |
| 16,2 | 119,7 | 18,4 | 109,9 |
| 16,4 | 118,8 | 18,6 | 109,0 |
| 16.6 | 117,9 | 18,8 | 108,1 |
| 16,8 | 117,0 | 19,0 | 107,2 |
| 17,0 | 116,1 | 19,2 | 106,3 |
| 17,2 | 115,2 | 19,4 | 105,4 |
| 17,4 | 114,3 | 19,6 | 104,5 |
| 17,6 | 113,4 | 19,8 | 103,7 |
15. DETERMINACIÓN DEL GRADO DE SEDIMENTACIÓN
(Según Zeleny)
15.1 Principio.
El esponjamiento de la fracción de gluten de harina en solución de ácido láctico afecta al grado de sedimentación de una suspensión de harina en el medio de ácido láctico. Más alto contenido en gluten y mejor calidad de éste, conduce a sedimentación más lenta y a más altos valores de la prueba de sedimentación.
El grado de sedimentación de una harina suspendida en una solución de ácido láctico durante un intervalo de tiempo estándar se toma como medida de su calidad panadera.
Es aplicable a harina de trigo.
15.2 Material y aparatos.
15.2.1 Pipeta de 25 ml
15.2.2 Pipeta de 50 ml
15.2.3 Cilindro graduado de 100 ml de vidrio o teflón, preferiblemente hecho de vidrio de precisión con una distancia de 180-185 mm, entre la marca cero y la marca 100 ml.
15.2.4 Reloj avisador o medida de intervalos de tiempo.
15.2.5 Bastidor mezclador movido por motor (agitador de vaivén).
El bastidor es aproximadamente de 58 × 32 × 5 cm. Se coloca en el centro de cada extremo y oscila 30° a cada lado de la horizontal y a una velocidad de 40 oscilaciones por minuto. El bastidor está diseñado para sujetar ocho cilindros, los cuales pueden ser colocados en sus posiciones rápidamente y con seguridad mientras el mezclador está en movimiento.
15.3 Reactivos.
15.3.1 Alcohol isopropílico, 99-100 por 100.
15.3.2 Agua destilada o desionizada. El agua utilizada para preparar los reactivos y el agua de hidratación no debe contener más de 2 p.p.m. de materia mineral.
15.3.3 Agua de hidratación (solución de azul de bromofenol). Añadir azul de bromofenol al agua destilada para conseguir una concentración de 4 mg por litro.
15.3.4 Solución de ácido láctico.
Diluir 250 ml de ácido láctico reactivo del 85 por 100 a 1 litro con agua destilada. Tener a reflujo el ácido diluido durante 6 horas sin pérdida de agua (15.6.2).
15.3.5 Reactivo de prueba de sedimentación. Mezclar íntimamente 180 ml de la solución preparada de ácido láctico (15.3.4) con 200 ml de alcohol isopropílico (15.3.1) y agua destilada hasta 1 litro. Dejar reposar durante 48 horas. Estandarizar a 0,050 ± 0,01 N utilizando hidróxido sódico o potásico 0,10 N. El peso específico debe ser 0,985 ± 0,001 a 15/15 °C. Evitar la evaporación.
La precisión debe ser como máximo de dos unidades.
15.4 Procedimiento.
Pesar 3,2 g de harina (14 por 100 humedad) y colocarla en un cilindro graduado y taponado. Añadir 50 ml de agua de hidratación con azul de bromofenol (15.3.3) y empezar a medir el tiempo al mismo tiempo. Mezclar harina y reactivo íntimamente sujetando el cilindro taponado en posición horizontal y agitando a derecha e izquierda a una distancia de 18 cm doce veces en cada dirección, cada 5 segundos. La harina debe estar completamente suspendida durante esta operación. Colocar el cilindro en el bastidor de mezcla y mezclar hasta que el tiempo transcurrido sea de 5 minutos. Quitar el cilindro del bastidor de mezcla y añadir 25 ml del reactivo (15.3.5). Volver el cilindro al bastidor hasta que el tiempo transcurrido sea de 10 minutos. Quitar el cilindro del bastidor y dejarlo reposar exactamente 5 minutos. Transcurridos estos 5 minutos exactos, leer el volumen del sedimento en ml estimando con precisión de 1/10 mililitros. Este es el grado de sedimentación.
15.5 Cálculo.
El grado de sedimentación es el anteriormente hallado. Los valores de sedimentación serán desde 8 aproximadamente para tipos de gluten con muy bajo contenido en proteína hasta 78 aproximadamente para tipo de gluten fuerte con muy alto contenido en proteína.
15.6 Observaciones.
15.6.1 El método descrito dará resultados concordantes cuan-do las harinas son producidas de la misma manera. Cuando empleamos muestras de trigo, los resultados dependen en gran manera del método de producción de harina utilizado. En general, los métodos de molienda que envuelven trituración con cilindros ondulados darán resultados comparables y situarán series de muestras en orden similar. Otros molinos, como el tipo de café, pueden dar resultados esencialmente carentes de significado.
15.6.2 El ácido láctico concentrado contiene moléculas asociadas, las cuales se disocian en disolución. Para resultados consistentes, la solución preparada de ácido láctico (15.3.4) debe haber alcanzado el equilibrio antes de su uso en el ensayo.
Esto se consigue por reflujo y almacenaje a temperatura ambiente.
15.6.3 Ambos, ácido láctico y alcohol isopropílico, deben estar esencialmente libres de materia mineral (no más de 40 p.p.m.).
15.6.4 La medida de los 5 minutos en el procedimiento es crítica y la lectura del grado de sedimentación debe ser hecha exactamente a los 5 minutos de reposo.
15.7 Referencia.
1. International Association for Cereal Chemistry (1. C. C.). Standard Nv. 116.
16. ÁCIDO ASCÓRBICO (Vitamina C)
(Método cualitativo)
16.1 Principio.
Este método sirve para determinar la presencia de vitamina C en harina y consiste en la aparición de puntos blancos sobre fondo azul del reactivo 2,6 diclorofenol indofenol.
16.2 Material y aparatos.
16.2.1 Placa Petri de 65 cm2.
16.3 Reactivos.
16.3.1 Disolución acuosa al 0,05 por 100 de 2,6 diclorofenol indofenol.
16.3.2 Disolución acuosa al 5 por 100 de ácido metafosfórico.
16.4 Procedimiento.
Extender 10 g de la muestra en la parte posterior de una cápsula Petri. Rociarla con la disolución de ácido metafosfórico y a continuación hacer lo mismo con la disolución del 2,6 diclorofenol indofenol. Al cabo de unos minutos aparecen unos puntos blancos más o menos grandes sobre el fondo azul-violáceo.
17. PERSULFATO AMÓNICO
(Método cualitativo)
17.1 Principio.
Este método sirve para determinar la presencia de persulfato amónico en harina y consiste en la aparición de manchas azules con la bencidina.
17.2 Material y aparatos.
17.2.1 Placa Petri de 65 cm2.
17.3 Reactivos.
17.3.1 Disolución de bencidina en etanol al 1 por 100 (p/v).
17.4 Procedimiento.
Extender 6 g de la muestra en la cápsula Petri. Añadir unos 3 ml de reactivo. Al cabo de unos minutos aparecen unas manchas azules.
Téngase en cuenta que la Orden de 26 de enero de 1989 Ref. BOE-A-1989-2695, por la que se aprueban los métodos oficiales de análisis de determinados tipos de leche parcial o totalmente deshidratada destinados a la alimentación humana, deroga los métodos coincidentes que figuran en la presente Orden.
Leche
1(a). GRASA
(Leches natural, certificada, higienizada y esterilizada)
1(a).1 Principio.
Este método es aplicable a las leches naturales, certificada, higienizada y esterilizada, enteras o parcialmente desnatadas, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de las leches natural, certificada, higienizada y esterilizada, el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-1A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente, por extracción de la citada materia grasa de una solución alcohólico-amoniacal del tipo de leche de que se trate, mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método de Rase-Gottlieb.
1(a).2 Material y aparatos.
1(a).2.1 Balanza analítica.
1(a).2.2 Probetas o matraces de extracción adecuados, provistos de tapones de vidrio esmerilado, de tapones de corcho u otros dispositivos de cierre inatacables por los disolventes utilizados. Los tapones de corcho serán de buena calidad y se tratarán sometiéndolos sucesivamente a extracciones con éter dietílico y con éter de petróleo. Después se introducirán durante 20 minutos, por lo menos, en agua a una temperatura de 60 °C o superior, y se dejarán enfriar también en agua, con objeto de que estén saturados cuando se utilicen.
1(a).2.3 Matraces de paredes delgadas y bases planas, de una capacidad de 150 a 250 ml.
1(a).2.4 Estufa de desecación, bien ventilada y controlada termostáticamente (ajustada para que funcione a una temperatura de 102 ± 2 °C), o una estufa de desecación por vacío (temperatura 70-75 °C, presión menor de 50 mm de Hg).
1(a).2.5 Materiales para facilitar la ebullición, exentos de materia grasa, no porosos ni deleznables al ser utilizados, como, por ejemplo, perlas de vidrio o trozos de carburo de silicio (el empleo de estos materiales es facultativo, véase el apartado 1(a).4.3).
1(a).3 Reactivos.
Todos los reactivos deberán ser de calidad pura para análisis y no dejar en la evaporación mayor cantidad de residuos que la autorizada para el ensayo en blanco (1(a).4.2). En caso necesario, los reactivos podrán destilarse de nuevo en presencia de 1 g aproximadamente de mantequilla deshidratada por 100 mililitros de disolvente. El agua que se utilice deberá ser destilada o, por lo menos, de igual pureza que el agua destilada.
1(a).3.1 Solución de amoníaco, de un 25 por 100 aproximadamente, en masa por volumen, de NH3 (densidad aproximada a 20 °C, 0,91 g por ml), o una solución más concentrada conociéndose dicha concentración.
1(a).3.2 Etanol de 96 ± 2 por 100 en volumen o, en su defecto, etanol desnaturalizado con metanol, etil-metil-cetona, benceno o éter de petróleo.
1(a).3.3 Éter dietílico, exento de peróxidos.
Para el ensayo de los peróxidos, añadir a 10 ml de éter dietilico contenidos en una pequeña probeta con tapón de vidrio, previamente enjuagada con un poco de éter, 1 ml de solución al 10 por 100 de yoduro potásico, recién preparada. Agitar y dejar reposar durante 1 minuto. No debe aparecer coloración amarilla en ninguna de las dos capas.
El éter dietílico puede mantenerse exento de peróxidos, añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante 1 minuto y después lavarse con agua. Por litro de éter dietílico, utilizar una superficie de 80 cm2 aproximadamente de lámina de cinc cortada en bandas lo suficientemente largas para que lleguen, por lo menos, hasta el centro del recipiente.
1(a).3.4 Éter de petróleo, de puntos de ebullición entre 30° y 60 °C.
1(a).3.5 Disolvente mixto, preparado poco tiempo antes de su utilización, mezclando volúmenes iguales de éter dietílico y éter de petróleo (se podrá sustituir la mezcla de disolventes en aquellos casos en que su utilización esté prevista por éter dietílico o por éter de petróleo).
1(a).4 Procedimiento.
1(a).4.1 Preparación de la muestra.‒Poner la muestra a una temperatura de 20 °C. Mezclarla cuidadosamente hasta obtener una distribución homogénea de la materia grasa. No agitar muy enérgicamente para evitar la formación de espuma en la leche o el batido de la materia grasa.
Si resulta dificultoso dispersar la capa de nata, calentar lentamente hasta 35-40 °C, mezclando cuidadosamente y teniendo la precaución de reincorporar a la muestra la nata que pudiera haberse adherido a las paredes del recipiente. Enfriar rápidamente la muestra hasta la temperatura ambiente. Si se desea se puede utilizar un homogeneizador apropiado para facilitar la dispersión de la grasa.
Si se separa la materia grasa líquida o se observa la presencia de partículas blancas de forma irregular adheridas a las paredes del recipiente que contiene la muestra, el análisis no dará resultados correctos.
1(a).4.2 Ensayo en blanco.‒Al mismo tiempo que se determina el contenido en materia grasa de la muestra, efectuar un ensayo en blanco con 10 ml de agua destilada en lugar de la muestra, empleando el mismo tipo de aparato de extracción, los mismos reactivos en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación. Si el resultado del ensayo en blanco excede de 0,5 mg, habrá que comprobar los reactivos, y aquel o aquellos que resulten impuros deberán sustituirse o purificarse.
1(a).4.3 Determinación.‒Secar el matraz [si se desea con algún material 1(a).2.5, que facilite una ebullición moderada durante la subsiguiente eliminación de los disolventes] en la estufa durante un intervalo de media a una hora. Dejar que se enfríe el matraz hasta la temperatura ambiente de la balanza y, una vez enfriado, pesarlo con una aproximación de 0,1 mg.
Invertir tres o cuatro veces el recipiente que contiene la muestra preparada y pesar inmediatamente en el aparato de extracción, directamente o por diferencia de 10 a 11 g de la muestra bien mezclada, con una aproximación de 1 mg. Añadir 1,5 ml de la solución de amoníaco al 25 por 100, o un volumen equivalente de una solución más concentrada, y mezclar convenientemente. Añadir 10 ml de etanol y mezclar suavemente, pero de modo homogéneo, manteniendo abierto el aparato de extracción. Añadir 25 ml de éter dietílico, cerrar el aparato y agitarlo vigorosamente, invirtiéndolo varias veces, durante 1 minuto. Si es necesario, enfriar el aparato con agua corriente. Quitar el tapón cuidadosamente y añadir 25 ml de éter de petróleo, utilizando los primeros ml para enjuagar el tapón y el interior del cuello del aparato, dejando que los líquidos de los enjuagues penetren en el último. Cerrarlo, volviendo a colocar el tapón, y agitarlo e invertirlo repetidamente durante 30 segundos. Si no está previsto centrifugar, en la operación descrita, no agitar muy enérgicamente.
Dejar el aparato en reposo hasta que la capa líquida superior esté completamente límpida y claramente separada de la fase acuosa. Podrá efectuarse igualmente la separación mediante el uso de una centrífuga adecuada Si se utiliza una centrífuga cuyo motor no sea trifásico, pueden producirse chispas y será preciso tomar las debidas precauciones para evitar una explosión o un incendio debido a la presencia de vapores de éter (por ejemplo, en caso de rotura de un tubo).
Quitar el tapón y enjuagarlo, así como también el interior del cuello del aparato, con algunos ml de la mezcla de disolventes, y dejar que los líquidos de los enjuagues penetren en el aparato. Transvasar con cuidado el matraz, lo más completamente posible, la capa superior de decantación o con la ayuda de un sifón. Si el transvase no se efectúa mediante sifón, tal vez sea necesario añadir un poco de agua para elevar la separación entre las dos capas, con objeto de facilitar la decantación.
Enjuagar el exterior e interior del cuello del aparato, o el extremo y la parte inferior del sifón, con algunos ml de la mezcla de disolventes. Dejar deslizar los líquidos del enjuague del exterior del aparato dentro del matraz y los del interior del cuello y del sifón, dentro del aparato de extracción.
Proceder a una segunda extracción repitiendo las operaciones descritas, desde la adición del éter dietílico, pero utilizando sólo 15 ml de éter dietílico y 15 ml de éter de petróleo. Efectuar una tercera extracción procediendo como se indica anteriormente, pero omitiendo el enjuague final. Eliminar con cuidado por evaporación o destilación la mayor cantidad posible de disolvente (incluido el etanol). Si el matraz es de pequeña capacidad será necesario eliminar un poco de disolvente después de cada extracción de la manera antes indicada.
Cuando ya no subsista olor a disolvente, calentar el matraz, tumbado, durante 1 hora en la estufa. Dejar que el matraz se enfríe hasta la temperatura ambiente de la balanza como se indicó y pesar con una aproximación de 0,1 mg. Repetir la operación calentando a intervalos de 30 y 60 minutos hasta obtener una masa constante. Añadir de 15 a 25 ml de éter de petróleo para comprobar si la materia extraída es totalmente soluble. Calentar ligeramente y agitar el disolvente mediante un movimiento rotatorio hasta que toda la materia grasa se disuelva.
Si la materia extraída es totalmente soluble en el éter de petróleo, la masa de materia grasa será la diferencia entre las pesadas efectuadas. En caso contrario o de duda, extraer completamente la materia grasa contenida en el matraz, mediante lavados repetidos con éter de petróleo caliente, dejando que se deposite la materia no disuelta antes de cada decantación. Enjuagar tres veces el exterior del cuello del matraz. Calentar el matraz, tumbado, durante 1 hora en la estufa y dejar que se enfríe hasta la temperatura ambiente de la balanza, como lo indicado anteriormente, y pesar con una aproximación de 0,1 miligramo. La masa de la Materia grasa será la diferencia entre la masa obtenida y la obtenida en esta pesada final.
1(a).5 Cálculo.
La masa, expresada en gramos, de la materia grasa extraída es:
(M1 ‒ M2) ‒ (B1 ‒ B2)
y el contenido en materia grasa de la muestra, expresado en porcentaje de la masa, es:

M1 = masa, en gramos, del matraz M, que contiene la materia grasa después de desecar hasta masa constante.
M2 = masa, en gramos, del matraz M, sin materia grasa, o en el caso de presencia de materias insolubles, después de extraer completamente la materia grasa.
B1 = masa, en gramos, del matraz B, del ensayo en blanco, después de desecar hasta masa constante.
B2 = masa, en gramos, del matraz B, o en el caso de presencia de materias insolubles, después de extraer completamente la materia grasa.
S = masa, en gramos, de la cantidad de muestra utilizada en la determinación.
La diferencia entre los resultados en dos determinaciones repetidas (resultados obtenidos simultáneamente o uno inmediatamente después de otro, por el mismo analista) no debe ser mayor de 0,03 g de materia grasa de 100 g de producto.
1(a).6 Referencia.
1. Norma internacional FIL-IDF 1A: 1969.
1(b). GRASA
(Leche desnatada)
1(b).1 Principio.
Este método es aplicable a las leches líquidas, no concentradas, y concretamente a la leche esterilizada desnatada, definida en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de la leche desnatada el porcentaje en masa de la cantidad total de lípidos y sustancias lipoides, determinada por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-22: 1963 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por adición de amoníaco y alcohol a una cantidad conocida de leche desnatada, extracción por un disolvente de los lípidos y de las sustancias lipoides, evaporación del disolvente y pesado del residuo, obtenido por aplicación del método Röse-Gottlieb.
1(b).2 Material y aparatos.
1(b).2.1 Balanza analítica, de sensibilidad mínima 0,1 miligramos.
1(b).2.2 Probetas o matraces de extracción apropiados provistos de tapones para cierre hermético (corcho o vidrio esmerilado).
1(b).2.3 Erlenmeyer o matraces de fondo plano, de 150 a 250 ml de capacidad, con zona esmerilada para identificación.
1(b).2.4 Materiales que faciliten la ebullición, exentos de materia grasa; por ejemplo, perlas de vidrio.
1(b).2.5 Dispositivos apropiados para la evaporación de los disolventes.
1(b).2.6 Estufa que permita obtener una temperatura constante de 102-104 °C o estufa de desecación por vacío.
1(b).2.7 Pipetas graduadas de 10 ml.
1(b).3 Reactivos.
1(b).3.1 Etanol del 96 ± 1 por 100 en volumen o etanol desnaturalizado con metanol o con éter de petróleo.
1(b).3.2 Éter dietílico, de punto de ebullición entre 34 y 35 °C, exento de peróxidos,
1(b).3.3 Éter de petróleo, de puntos de ebullición entre 40 y 60 °C.
1(b).3.4 Solución de amoníaco del 25 por 100 (densidad 0,91 a 20 °C) límpida e incolora.
Los reactivos no deben dejar residuos después de la evaporación.
1(b).4 Procedimiento.
1(b).4.1 Preparación de la muestra.‒Mezclar la muestra cuidadosamente. En caso necesario, calentar la muestra a 35-40 °C y mezclar. Enfriar a 20 °C antes del análisis.
1(b).4.2 Determinación.‒Con una aproximación de 10 miligramos, pesar alrededor de 10 g, o introducir exactamente 10 milímetros (a 20 ± 2 °C) de leche desnatada en el aparato de extracción. Añadir 1 ml de la solución de amoníaco y mezclar cuidadosamente. Añadir 10 ml de etanol y mezclar el contenido. Añadir 25 ml de éter dietílico y, después de cerrar el aparato de extracción, mezclar el contenido agitando e invirtiendo varias veces durante 1 minuto. Añadir 25 ml de éter de petróleo, cerrar el aparato de extracción y mezclar el contenido agitando e invirtiendo repetidas veces.
Dejar reposar el aparato de extracción por lo menos 2 horas o centrifugar (por lo menos 5 minutos a 500-600 revoluciones por minuto) hasta que la capa éter dietílico-éter de petróleo esté totalmente límpida y completamente separada de la fase acuosa. Transvasar, lo más completamente posible, la capa éter dietílico-éter de petróleo por decantación, o con ayuda de un dispositivo de sifón (teniendo cuidado de no traspasar la fase acuosa), a un erlenmeyer o matraz de fondo plano, que contenga un material destinado a facilitar la ebullición, previamente desecado y pesado.
Realizar una segunda extracción utilizando 15 ml de éter dietílico y 15 ml de éter de petróleo, siguiendo el procedimiento indicado. Transvasar al mismo matraz la capa éter dietílico-éter de petróleo como se indica anteriormente. Destilar con cuidado los disolventes contenidos en el matraz.
Después de la evaporación de los disolventes, secar la materia grasa durante 1 hora en estufa de vacío a 70°-75 °C (presión inferior a 50 mm de mercurio), o en una estufa de presión normal a 102-104 °C, colocando el matraz en posición horizontal. Dejar enfriar el matraz y pesar cuando haya alcanzado la temperatura ambiente. Continuar el proceso de desecación hasta peso constante (desecación en vacío) o hasta un ligero aumento del peso (desecación a presión normal). En el último caso, tomar para el cálculo el último valor encontrado antes del aumento del peso. Si se considera necesario, la materia grasa puede volver a disolverse en éter de petróleo para comprobar el resultado del análisis.
1(b).4.3 Ensayo en blanco.‒Para el control de los reactivos es necesario efectuar un análisis en blanco siguiendo exactamente la forma de operar indicada y utilizando 10 ml de agua en lugar de leche desnatada. El ensaya en blanco no deberá indicar más que una cantidad inapreciable.
Para determinar la influencia de las variaciones de temperatura y humedad del aire, pesar un matraz y tratarlo como el utilizado para el análisis, pero sin llenarlo con los disolventes.
1(b).5 Cálculo.
En el cálculo de porcentaje de materia grasa se tendrán en cuenta, si es necesario, los valores encontrados en los ensayos en blanco. Si la cantidad de leche tomada para el análisis se ha tomado con ayuda de una pipeta, es necesario tener en cuenta la densidad de la leche.
La diferencia entre los resultados en dos determinaciones repetidas no debe ser mayor de 0,005 g de materia grasa por 100 g de producto.
1(b).6 Referencia.
1. Norma Internacional FIL-IDF 22: 1963.
1(c). GRASA
(Leches concentrada, evaporada y condensada)
1(c).1 Principio.
Este método es aplicable a las leches concentrada, evaporada y condensada, enteras o desnatadas, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de las leches concentrada, evaporada y condensada el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-13A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por extracción de la citada materia grasa en una solución alcohólico-amoniacal del tipo de leche de que se trate, mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método de Röse-Gottlieb.
1(c).2 Material y aparatos.
1(c).2.1 Balanza analítica.
1(c).2.2 Probetas o matraces de extracción adecuados provistos de tapones de vidrio esmerilado, de tapones de corcho u otros dispositivos de cierre inatacables por los disolventes utilizados. Los tapones de corcho serán de buena calidad y se tratarán sometiéndolos sucesivamente a extracciones con éter dietílico y con éter de petróleo. Después se introducirán, durante 20 minutos por lo menos, en agua a una temperatura de 60 °C o superior y se dejarán enfriar también en agua con objeto de que estén saturados cuando se utilicen.
1(c).2.3 Matraces de paredes delgadas y bases planas de una capacidad de 150 a 250 ml.
1(c).2.4 Estufa de desecación bien ventilada y controlada termostáticamente (ajustada para que funcione a una temperatura de 102 ± 2 °C) o una estufa de desecación por vacío (temperatura 70-75 °C, presión menor de 50 mm de Hg).
1(c).2.5 Materiales para facilitar la ebullición, exentos de materia grasa, no porosos ni deleznables al ser utilizados, como, por ejemplo, perlas de vidrio o trozos de carburo de silicio (el empleo de estos materiales es facultativo).
1(c).3 Reactivos.
Todos los reactivos deben ser de calidad pura para análisis y no dejar en la evaporación mayor cantidad de residuos que la autorizada para el ensayo en blanco. En caso necesario, los reactivos podrán destilarse de nuevo en presencia de 1 g, aproximadamente, de mantequilla deshidratada por 100 ml de disolvente. El agua que se utilice deberá ser destilada o por lo menos de igual pureza que el agua destilada.
1(c).3.1 Solución de amoníaco de un 25 por 100, aproximadamente, en masa por volumen de NH3 (densidad aproximada a 20 °C, 0,91 g por ml) o una solución más concentrada, conociéndose dicha concentración.
1(c).3.2 Etanol de 96 ± 2 por 100 en volumen, o etanol desnaturalizado con metanol, etil-metil-cetona, benceno o éter de petróleo.
1(c).3.3 Éter dietílico exento de peróxidos.
Para el ensayo de los peróxidos, añadir a 10 ml de éter dietílico contenidos en una pequeña probeta con tapón de vidrio, previamente enjuagada con un poco de éter, 1 ml de solución al 10 por 100 en yoduro potásico recién preparada. Agitar y dejar reposar durante un minuto. No debe aparecer coloración amarilla en ninguna de las dos capas.
El éter dietílico puede mantenerse exento de peróxidos añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante 1 minuto y después lavarse con agua. Por litro de éter dietílico utilizar una superficie de 80 cm2, aproximadamente, de lámina de cinc cortada en bandas lo suficientemente largas para que lleguen, por lo menos, hasta el centro del recipiente.
1(c).3.4 Éter de petróleo de puntos de ebullición entre 30 y 60 °C.
1(c).3.5 Disolvente mixto preparado poco tiempo antes de su utilización mezclando volúmenes iguales de éter dietílico y éter de petróleo (se podrá sustituir la mezcla de disolventes, en aquellos casos en que su utilización esté prevista, por éter dietílico o por éter de petróleo).
1(c).4 Procedimiento.
1(c).4.1 Preparación de la muestra.
1(c).4.1.1 Leche concentrada y leche evaporada.‒Agitar e invertir el recipiente que contiene la muestra. Abrir y transvasar lentamente la leche a un segundo recipiente (provisto de cierre hermético). Mezclar mediante transvases sucesivos, teniendo cuidado de incorporar a la muestra toda la materia grasa u otro constituyente adheridos a las paredes o al fondo del primer recipiente. Finalmente, transvasar la leche lo más completamente posible al segundo recipiente y cerrar este último.
En caso necesario, templar el envase original, cerrado, en baño de María a 40-60 °C. Sacarlo y agitar vigorosamente cada 15 minutos. Al cabo de 2 horas, retirar el envase y dejarlo enfriar hasta temperatura ambiente. Quitar totalmente la tapadera y mezclar cuidadosamente removiendo el contenido con una cuchara o espátula (si se separa la materia grasa no se debe efectuar el análisis de la muestra).
En el caso de envases flexibles, abrirlos y transvasar el contenido a un vaso. Rasgar los envases, despegar todas las materias adheridas a las paredes e introducirlas en el vaso.
1(c).4.1.2 Leche condensada.‒Abrir el recipiente que contiene la muestra y mezclar cuidadosamente la leche con una cuchara o una espátula. Imprimir a este instrumento un movimiento rotativo ascendente y descendente de manera que las capas superiores e inferiores se mezclen bien con el resto del contenido. Tener cuidado de reincorporar a la muestra toda la masa de leche que pudiera haberse adherido a las paredes y a los fondos del recipiente. Transvasar la leche lo más completamente posible a un segundo recipiente (provisto de cierre hermético) y cerrar este último.
En caso necesario, templar el bote original, cerrado, en baño de María a 30-40 °C. Abrir el bote, desprender toda la leche adherida a las paredes del mismo, transvasar a una cápsula lo suficientemente grande para permitir un manejo cuidadoso y mezclar hasta que toda la masa sea homogénea.
En el caso de tubos flexibles, abrirlos y transvasar el contenido a un vaso. Rasgar los tubos, despegar todas las materias adheridas a las paredes e introducirlas en el vaso.
1(c).4.2 Ensayo en blanco.‒Al mismo tiempo que se determina el contenido en materia grasa de la muestra, efectuar un ensayo en blanco con 10 ml de agua destilada en lugar de la muestra, empleando el mismo tipo de aparato de extracción, los mismos reactivos en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación. Si el resultado del ensayo en blanco excede de 0,5 mg, habrá que comprobar los reactivos y aquél o aquéllos que resulten impuros deberán sustituirse o purificarse.
1(c).4.3 Determinación.‒Proceder como en 1 (a).4.3, excepto que se pesa de 4 a 5 g de la muestra, añadiendo a continuación 7 ml de agua, agitando suavemente y calentando ligeramente (40-50 °C) hasta la dispersión total del producto.
1(c).5 Cálculo.
Como en 1(a).5.
1(c).6 Referencia.
1. Norma Internacional FIL-IDF 13A: 1969.
1(d). GRASA
(Leche en polvo)
1(d).1 Principio.
Este método es aplicable a las leches en polvo, entera, parcialmente desnatada y desnatada, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de la leche en polvo el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-9A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por extracción de la citada materia grasa de una solución alcohólico-amoniacal de leche en polvo mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método Röse-Gottlieb.
1(d).2 Material y aparatos.
1(d).2.1, 2, 3, 4, 5 y 6 como 1(a).2.1, 2, 3, 4, 5 y 6.
1(d).3 Reactivas.
1(d).3.1, 2, 3, 4 y 5, como 1(a).3.1, 2, 3, 4 y 5.
1(d).4 Procedimiento.
1(d).4.1 Preparación de la muestra.‒Transvasar la leche en polvo a un recipiente limpio y seco (provisto de cierre hermético) de una capacidad que corresponda, aproximadamente, a dos veces el volumen del polvo. Cerrar en seguida el recipiente y mezclar cuidadosamente la leche en polvo agitándolo e invirtiéndolo repetidamente. Durante la preparación de la muestra deberá evitarse, en la medida de lo posible, la exposición de la leche en polvo al aire atmosférico con objeto de reducir al mínimo la absorción de humedad.
1(d).4.2 Ensayo en blanco.‒Como en 1(a).4.2.
1(d).4.3 Determinación.‒Como en 1 (a).4.3, excepto que se pesa, aproximadamente, 1 g de leche entera en polvo o 1,5 g de leche parcialmente desnatada en polvo o de leche desnatada en polvo añadiendo 10 ml de agua y agitando hasta la dispersión total del polvo de leche. Después de añadir la solución de amoníaco, calentar en baño María a una temperatura de 60-70 °C durante 15 minutos, agitando de cuando en cuando, y enfriar a continuación con agua corriente, si se desea.
1(d).5 Cálculo.
Como en 1(a).5.
1(d).6 Referencia.
1. Norma Internacional FIL-IDF 9A: 1969.
1(e). GRASA
(Leche natural) (Método Gerber)
En el caso de leche natural se puede emplear, como alternativo, el método Gerber.
1(e).1 Principio.
Liberación total de la grasa por disolución de las sustancias proteicas, separación de la grasa por centrifugación y posterior medida volumétrica de ésta.
Aplicable a leche natural, pasterizada y esterilizada.
1(e).2 Material y aparatos.
1(e).2.1 Pipetas aforadas de 11 ml.
1(e).2.2 Dosificador de émbolo de 10 ml para el ácido sulfúrico.
1(e).2.3 Baño de agua regulable a 65 °C.
1(e).2.4 Centrífuga Gerber.
1(e).2.5 Butirómetros original Gerber. Se pueden utilizar de varios tipos.
1(e).2.5.1 Graduación de 0-5 por 100 divisiones en 0,1. Cuello ranurado.
1(e).2.5.2 Graduación de 0-5 por 100 divisiones en 0,1. Cuello liso.
1(e).2.5.3 Graduación de 0-7 por 100 divisiones en 0,1. Cuello ranurado.
1(e).2.5.4 Graduación de 0-9 por 100 divisiones en 0,1. Cuello ranurado.
1(e).2.6 Tapones de caucho.
1(e).2.7 Empujador metálico.
1(e).3 Reactivos.
1(e).3.1 Ácido sulfúrico d = 1,82 y riqueza 90-91 por 100.
1(e).3.2 Alcohol isoamílico.
1(e).4 Procedimiento.
Colocar en el butirómetro 10 ml de SO4H2 [1(e)3.1] y agregar 11 ml de leche con cuidado y lentamente para que no se mezclen, observándose claramente la separación de ambas capas, ácida y de leche.
Agregar a continuación 1 ml de alcohol isoamílico (con dosificador) y cerrar el butirómetro. Agitar enérgicamente, envuelto en un paño para evitar posibles proyecciones hasta la total disolución de la fase proteica de la leche. Verter y dejar en reposo algún tiempo para observar mejor si la disolución ha sido completa. Llevar a la centrífuga durante 5 minutos. Sacar de la centrífuga con cuidado para no mover la capa superior de grasa ya separada. Colocar en el bario (65 °C) durante 5 minutos. Sacar y leer rápidamente.
1(e).5 Cálculo.
Se lee directamente en la escala del butirómetro.
1(e).6 Observaciones.
1(e).6.1 A veces el volumen de líquidos en el butirómetro no es suficiente para que la columna de grasa quede en la escala graduada; en este caso, antes de centrifugar, añadir unas gotas de ácido sulfúrico o simplemente agua si es después de la centrifugación, para conseguir que dicha columna pueda ser leída.
1(e).6.2 Puede ser conveniente después de la agitación del butirómetro y antes de centrifugar, colocarlo en el baño a 65 °C de 5 a 15 minutos para que el ataque sea lo más completo posible, aunque presenta el inconveniente de hacer el glóbulo graso más pequeño, produciéndose un ligero aumento del volumen de grasa, obteniéndose un valor ligeramente por exceso. A pesar de todo, si dicho tiempo de resistencia en el baño no se prolonga más allá de esos 15 minutos, la variación apenas es apreciable, quedándose dentro del error experimental 0,05 por 100 en MG propio del método.
Sin embargo, presenta la ventaja de apreciar claramente antes de la centrifugación si la disolución ha sido total, factor fundamental en la determinación.
1(e).6.3 En leches conservadas mediante formol, la disolución de la proteína es imposible o incompleta, por lo que la separación de la capa grasa en la centrifugación no es completa ni nítida. No es aconsejable en dicho caso el empleo de este método.
2. PROTEÍNAS
2.1 Principio.
Se entiende por contenido en proteínas de la leche el contenido en nitrógeno expresado en porcentaje en peso y multiplicado por el factor de conversión, que se determina por el método expuesto a continuación, el cual corresponde al descrito en la norma FIL-20: 1962 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas, natural, certificada, higienizada, esterilizada y a las reconstituidas asimismo no alteradas, posteriormente de las leches concentrada, evaporada, condensada y en polvo.
La determinación del nitrógeno total se realiza por aplicación del método Kjeldahl: una determinada cantidad pesada de leche se trata con ácido sulfúrico en presencia de óxido de mercurio como catalizador con objeto de transformar el nitrógeno de los compuestos orgánicos en nitrógeno amoniacal. El amoníaco se libera por adición de sosa cáustica, se destila y se recoge a una solución de ácido bórico. A continuación se valora el amoníaco.
2.2 Material y aparatos.
2.2.1 Balanza analítica de 1 mg de sensibilidad mínima.
2.2.2 Aparato de digestión que permita mantener el matraz Kjeldahl en una posición inclinada y provisto de un sistema de calentamiento que no afecte más que a la parte del matraz ocupada por el líquido.
2.2.3 Matraz Kjeldahl de 500 ml de capacidad.
2.2.4 Refrigerante Liebig de tubo interior rectilíneo.
2.2.5 Un tubo de salida con bulbo de seguridad esférico, conectado a la parte inferior del refrigerante por unión esmerilada.
2.2.6 Una alargadera conectada al matraz Kjeldahl y al refrigerante Liebig por medio de goma. Uniones esmeriladas.
2.2.7 Un matraz erlenmeyer de 500 ml de capacidad.
2.2.8 Probetas graduadas de 25, 50, 100 y 150 ml.
2.2.9 Bureta de 50 ml graduados a 0,1 ml.
2.2.10 Materiales para facilitar la ebullición. En la digestión, pequeños trozos de porcelana dura o perlas de vidrio, y en la destilación, pequeños trozos de piedra pómez recién calcinados.
2.3 Reactivos.
2.3.1 Sulfato potásico.
2.3.2 Oxido de mercurio rojo.
2.3.3 Ácido sulfúrico concentrado (densidad: 1,84 a 20 °C).
2.3.4 Solución de sosa cáustica: 500 g de hidróxido sódico (NaOH) y 12 g de sulfuro de sodio (SNa2 ∙ 9H2O) disueltos en 1.000 ml de agua destilada.
2.3.5 Solución de ácido bórico: 40 g de ácido bórico disuelto en 1.000 ml de agua destilada.
2.3.6 Ácido clorhídrico 0,1 N.
2.3.7 Indicador: 2 g de rojo de metilo y 1 g de azul de metileno disueltos en 1.000 ml de alcohol etílico del 96 por 100.
2.3.8 Solución de tetraborato de sodio para la valoración del ácido clorhídrico.
Los reactivos y las soluciones utilizadas no deben contener sustancias nitrogenadas.
2.4 Procedimiento.
2.4.1 Preparación de la muestra.‒Antes del análisis poner la muestra a 20 ± 2 °C y mezclarla cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentarla lentamente a 40 °C, mezclar suavemente y enfriarla de nuevo a 20 ± 2 °C.
2.4.2 Determinación.‒Introducir sucesivamente en el matraz Kjeldahl algunas perlas de vidrio o pequeños trozos de porcelana, alrededor de 10 g de sulfato potásico, 0,5 g de óxido de mercurio y alrededor de 5 g de leche exactamente pesados, con aproximación de 1 mg.
Añadir 20 ml de ácido sulfúrico y mezclar el contenido del matraz. Calentar cuidadosamente el matraz Kjeldahl sobre el dispositivo para la reacción hasta que no se forme espuma y el contenido se vuelva líquido. Continuar la reacción por calentamiento más intenso, hasta que el contenido del matraz esté perfectamente límpido e incoloro. Durante el calentamiento, agitar de cuando en cuando el contenido del matraz, Cuando el líquido esté perfectamente límpido, proseguir la ebullición durante una hora y media, evitando todo sobrecalentamiento local.
Dejar enfriar el contenido del matraz a la temperatura ambiente, añadir alrededor de 150 ml de agua destilada y algunos fragmentos de piedra pómez, mezclar cuidadosamente y dejarlo todavía enfriar algo más, Con la ayuda de una probeta graduada, verter 50 ml de solución de ácido bórico en el matraz erlenmeyer, añadir cuatro gotas de indicador y mezclar. Situar el matraz erlenmeyer bajo el refrigerante, de manera que el extremo del tubo de salida se introduzca en la solución de ácido bórico. Con la ayuda de una probeta graduada añadir al con-tenido del matraz Kjeldahl 80 ml de la solución de sosa cáustica que contiene sulfuro. Durante esta operación, mantener el matraz inclinado, de tal manera que la sosa se deslice a lo largo de la pared del recipiente y que los líquidos no se mezclen. Conectar inmediatamente el matraz Kjeldahl al refrigerante por medio de la alargadera. Mezclar el contenido del matraz por agitación. Calentar a ebullición evitando la espuma. Proseguir la destilación hasta el momento en que el contenido del matraz presente ebullición a saltos. Regular el calentamiento de manera que la destilación dure por lo menos 20 minutos. Enfriar bien el destilado para evitar que se caliente la solución del ácido bórico. Poco tiempo antes de terminar la destilación, bajar el matraz erlenmeyer para que el tubo de salida no esté introducido en la solución de ácido bórico. Detener el calentamiento; elevar el tubo de salida y enjuagar sus paredes exteriores e interiores con un poco de agua destilada. Valorar el destilado con ácido clorhídrico 0,1 N.
2.4.3 Ensayo en blanco.‒Efectuar un ensayo en blanco, aplicando el método operatorio descrito, pero utilizando cinco mililitros de agua destilada en lugar de leche.
2.5 Cálculo.
2.5.1 Nitrógeno total.‒Se calcula el contenido en nitrógeno total mediante la fórmula

N = normalidad del ácido clorhídrico.
V1 = volumen en mililitros de ácido clorhídrico utilizado en la determinación.
V0 = volumen en mililitros de ácido clorhídrico utilizado en el ensayo en blanco
P = peso en gramos de la leche empleada en el análisis.
La diferencia máxima entre dos determinaciones repetidas no debe sobrepasar el 0,005 por 100 de nitrógeno.
2.5.2 Proteínas.‒Para expresar el contenido en proteínas de la leche analizada es preciso multiplicar la cantidad de nitrógeno total, obtenida según el método descrito, por un factor de conversión que se fija en 6,38. En consecuencia, el contenido en proteínas viene dado por la fórmula

2.6 Referencia.
1. Norma Internacional FIL-IDF 20: 1962.
3. CASEÍNA
3.1 Principio.
Se entiende por contenido en caseína de la leche el contenido en proteínas, expresado en porcentaje en peso, obtenidas después de una precipitación a pH 4,6, siguiendo el procedimiento expuesto a continuación, que corresponde a la norma FIL-29: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas, natural, certificada, higienizada, esterilizada y a las reconstituidas también, no alteradas posteriormente, de las leches concentrada, evaporada, condensada y en polvo. Asimismo puede aplicarse a las muestras de leche conservadas por adición de formaldehido (1: 2.500).
Se determina la cantidad total de nitrógeno de la leche. A continuación la caseína se precipita con un tampón acético-acetato y se filtra. Se determina luego la cantidad de nitrógeno del filtrado.
La cantidad de caseína se calcula con estas dos determinaciones de nitrógeno, que se realizan por el método Kjeldahl, según el método 2.
3.2 Material y aparatos.
3.2.1 Pipetas de 0,5, 1 y 10 ml.
3.2.2 Probeta graduada de 100 ml.
3.2.3 Matraz graduado de 100 ml.
3.2.4 Papel filtro, lavado en ácido, velocidad media: de 11 a 12,5 cm.
3.2.5 Embudos.
3.3 Reactivos.
3.3.1 Solución de ácido acético al 10 por 100 (en peso por volumen).
3.3.2 Solución de acetato de sodio, 1 N.
3.4 Procedimiento.
3.4.1 Preparación de la muestra.‒Antes del análisis poner la muestra a 20 ± 2 °C y mezclar cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentar la muestra lentamente a 40 °C; mezclar suavemente y enfriar a 20° ± 2 °C.
3.4.2 Determinación.‒Determinar el contenido total en nitrógeno (NT) de la leche utilizando el método Kjeldahl, según el método 2.
Precipitar la caseína de la siguiente manera:
Llevar con pipeta 10 ml de leche a un matraz aforado de 100 ml. Añadir 75 ml de agua a 40 °C; después un ml de la solución de ácido acético. Mezclar suavemente el contenido del matraz y esperar 10 minutos. Añadir con pipeta un ml de la solución de acetato de sodio. Mezclar de nuevo. Dejar enfriar el contenido del matraz a unos 20 °C, completar hasta 10 ml con agua y mezclar invirtiendo lentamente el matraz. Cuando el precipitado de caseína y materia grasa se haya depositado, filtrar con filtro seco y recoger el filtrado en un recipiente seco.
Determinar el contenido en nitrógeno de 50 ml del filtrado límpido (nitrógeno no caseínico: NNC), utilizando el método Kjeldahl según el método 2.
3.4.3 Ensayo en blanco.‒Además del ensayo en blanco previsto en el método 2, relativo a la determinación del contenido en nitrógeno total de la leche, conviene hacer un ensayo en blanco para comprobar los reactivos que precipitan la caseína, utilizando 50 ml de agua, 0,5 ml de la solución de ácido acético y 0,5 ml de la solución de acetato de sodio.
3.5 Cálculo.
Calcular el porcentaje en peso de NT en la leche y del NNC en el suero límpido con tres cifras significativas. Corregir la cifra obtenida para NNC por el volumen del precipitado, multiplicando el valor obtenido por 0,994. Calcular la cantidad de caseína mediante la fórmula
Cantidad de caseína % = 6,38 (NT — NNC)
La aplicación de este factor a todas las leches enteras no entraña error sensible; sin embargo, si se aplica el método a leche desnatada, el factor de corrección utilizado deberá ser 0,998.
La diferencia entre dos determinaciones repetidas no debe sobrepasar el 0,04 por 100 de caseína.
3.6 Referencia.
1. Norma Internacional FIL-IDF 29: 1964.
4. LACTOSA
4.1 Principio.
Se entiende por contenido en lactosa de la leche el contenido en lactosa monohidratada expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-28: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas natural, certificada, higienizada, esterilizada y a las reconstituidas, asimismo no alteradas posteriormente, de las leches concentrada, evaporada, condensada y en polvo. También puede aplicarse a las muestras de leche conservadas por adición de formaldehido (1:2.500).
El contenido en lactosa se determina indirectamente, una vez desproteinizada la leche, por valoración de la cantidad de halógeno reducido al final de la reducción entre lactosa y yoduro potásico-cloramina T.
4.2 Material y aparatos.
4.2.1 Balanza analítica.
4.2.2 Matraces aforados de 100 ml.
4.2.3 Pipetas de 5, 10, 20, 25 y 40 ml.
4.2.4 Papel filtro (lavado en ácido, velocidad media: 11 a 12,5 cm).
4.2.5 Embudos filtrantes.
4.2.6 Matraces erlenmeyer, de una capacidad de 150 a 200 ml.
4.2.7 Matraces erlenmeyer, de 150 ml con cierre esmerilado.
4.2.8 Bureta de 10 ml graduada a 0,02 ml.
4.3 Reactivos.
4.3.1 Reactivo del ácido tungsténico: Disolver 7 g de tungstanoto de sodio (WO4Na2 ∙ 2H2O) en 870 ml de agua; añadir 0,1 ml de una solución de ácido ortofosfórico (88 por 100 en peso) y 70 ml de una solución de ácido sulfúrico 1 N.
4.3.2 Solución de cloramina T, 0,040 N (5,70 g por litro).
4.3.3 Solución de tiosulfato de sodio normalizada a un poco más de 0,040 N.
4.3.4 Solución de yoduro potásico del 10 por 100, recientemente preparada, incolora.
4.3.5 Solución de ácido clorhídrico, 2 N.
4.3.6 Solución de almidón soluble del 1 por 100.
4.4 Procedimiento.
4.4.1 Preparación de la muestra.‒Antes de análisis poner la muestra a 20 ± 2 °C y mezclarla con cuidado. Si no se obtiene una dispersión homogénea de la materia grasa, calentar la muestra lentamente a 40 °C, mezclar suavemente y enfriar a 20 ± 2 °C.
4.4.2 Determinación.‒Llevar con pipeta 10 ml de leche a matraz aforado de 100 ml. Añadir 25 ml de agua, 40 ml del reactivo de ácido tungsténico y mezclar suavemente. Completar hasta 100 ml con agua, mezclar y dejar que se deposite el precipitado. Filtrar con un filtro seco y recoger en un matraz seco. Con una pipeta tomar 10 ml de filtrado y ponerlo en un matraz erlenmeyer de 150 ml provisto de tapón esmerilado. Añadir 5 ml de la solución de yoduro potásico y exactamente 20 ml de la solución de cloramina T. Mezclar. Tapar el matraz con su tapón, previamente humedecido con un poco de solución de yoduro potásico y mantenerlo en la oscuridad durante una hora y media a 18-20 °C. Quitar el tapón, enjuagarlo en el matraz con un poco de agua y añadir 5 ml de la solución de ácido clorhídrico. Añadir exactamente 10 ml de la solución de tiosulfato de sodio. Valora con una aproximación de 0,02 ml con la solución de tiosulfato. Hacia el final de la valoración, añadir dos o tres gotas de la solución de almidón.
4.4.3 Ensayo en blanco.‒Efectuar un ensayo en blanco siguiendo exactamente el método operatorio descrito, pero empleando 10 ml de agua en lugar del filtrado.
4.5 Cálculo.
Calcular la diferencia entre los valores de tiosulfato obtenidos para el ensayo en blanco y para el de la leche. Corregir el valor en función del volumen del precipitado, multiplicando por 0,992. Convertir la cifra obtenida en lactosa monohidratada, teniendo en cuenta que 1 ml de solución de tiosulfato 0,040 N corresponde a 0,00720 g de lactosa monohidratada. Expresar los resultados en porcentajes de lactosa monohidratada.
La aplicación de este factor 0,992 a todas las leches enteras no ocasiona ningún error sensible; sin embargo, si el método se aplica a la leche desnatada, debe utilizarse 0,996 como factor de corrección.
La diferencia máxima entre dos determinaciones repetidas no debe sobrepasar de 0,05 por 100 de lactosa.
4.6 Observaciones.
La solución de tiosulfato se debe normalizar periódicamente, por ejemplo, por valoración de 10 ml de yodato de potasio 0,040 N, a los que se habrá añadido 5 ml de la solución de yoduro potásico y 5 ml de la solución de ácido clorhídrico.
La concentración de la solución de tiosulfato ha sido elegida de tal forma que la lectura en bureta sea de 2 a 3,5 ml para la mayor parte de las muestras de leche y de 9,5 a 9,7 ml para los ensayos en blanco.
Cuando se proceda a una serie de análisis se deben preparar los filtrados y los ensayos en blanco hasta la adición de yoduro de potasio inclusive. Finalmente se añade rápidamente la solución de cloramina T a cada matraz y se anota la hora. Después de una hora y media (se admite una tolerancia de una hora veinte minutos a una hora cuarenta minutos) se añade el ácido clorhídrico en todos los matraces y siguiendo el mismo orden. Así se paraliza la reacción y los matraces se pueden valorar por turno
4.7 Referencia.
1. Norma Internacional FIL-IDF 28: 1967.
5(a). EXTRACTO SECO
(Leches natural, certificada, higienizada y esterilizada)
5(a).1 Principio.
Se entiende por contenido en extracto seco de las leches natural, certificada, higienizada y esterilizada, el residuo, expresado en porcentaje en peso, obtenida después de efectuada la desecación de la leche de que se trate por el procedimiento expuesto a continuación, que corresponde al descrito en la norma F1L-21: 1962 de la Federación Internacional de Lechería.
Una cantidad conocida de leche se deseca a temperatura constante hasta peso constante. El peso obtenido después de desecar representa el de la materia seca.
5(a).2 Material y aparatos.
5(a).2.1 Balanza analítica, de sensibilidad 0,1 ml como mínimo.
5(a).2.2 Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
5(a).2.3 Estufa de desecación que permita conseguir una temperatura constante de 102 ± 2 °C.
5(a).2.4 Cápsulas metálicas planas, en metal inoxidable o de vidrio de 2 cm de altura aproximadamente y de 6 a 8 cm de diámetro, aproximadamente, con tapas adecuadas.
5(a).2.5 Baño de agua.
5(a).3 Procedimiento.
5(a).3.1 Preparación de la muestra.‒Antes del análisis, poner la muestra a 20 ± 2 °C y mezclarla cuidadosamente. Si no se obtiene una buena repartición de la materia grasa, calentar lentamente a 40 °C, mezclarla suavemente y enfriarla, a 20 ± 2 °C.
5(a).3.2 Determinación.‒Secar la cápsula y la tapa a 102 ± 2 °C durante 30 minutos. Colocar la cápsula tapada en un desecador, dejarla enfriar a la temperatura ambiente y pesarla. Poner aproximadamente 3 ml de la muestra en la cápsula, tapar la cápsula con la tapa y pesarla. Poner la cápsula destapada en baño de agua durante 30 minutos. Poner la cápsula y la tapa en una estufa de desecación a 102 ± 2 °C durante 2 horas. La tapa se debe poner a un lado de la cápsula. Cubrir la cápsula con la tapa y ponerla en el desecador; dejarla enfriar y pesarla. Ponerla 1 hora más en la estufa de desecación; dejarla enfriar y pesarla. Repetir la desecación hasta que la diferencia entre dos pesadas consecutivas no sea mayor de 0,5 mg.
5(a).4 Cálculo.

P' = peso en gramos de la muestra después de la desecación.
P = peso en gramos de la muestra antes de la desecación.
Si a la muestra de la leche se le han adicionado como conservadores sustancias no volátiles, como, por ejemplo, dicromato potásico, se debe corregir la expresión del extracto seco como sigue:

C' = cantidad de conservante no volátil en la muestra analizada.

En caso de ser dicromato potásico, el contenido porcentual de conservante se determina según el método 7.
La diferencia entre dos determinaciones repetidas no debe ser mayor de 0,05 por 100 del extracto seco.
5(a).5 Referencia.
1. Norma Internacional FIL-IDF 21: 1962.
5(b). EXTRACTO SECO
(Leches concentrada, evaporada y condensada)
5(b).1. Principio.
Se entiende por contenido en extracto seco de las leches concentrada, evaporada y condensada, el residuo, expresado en porcentaje en peso obtenido después de efectuada la desecación de la leche de que se trate, por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-15: 1961 de la Federación Internacional de Lechería.
El método consiste en la dilución con agua de una cantidad conocida de muestra y la desecación con arena a temperatura constante. El peso obtenido después de desecar representa el de la materia seca.
5(b).2 Material y aparatos.
5(b).2.1 Balanza analítica de sensibilidad: 0,1 mg como mínimo.
5(b).2.2 Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
5(b).2.3 Estufa de desecación que permita obtener una temperatura constante de 98-100 °C.
5(b).2.4 Cápsulas metálicas, planas, en metal inoxidable o de vidrio de 2,5 cm de altura aproximadamente, y de 7,5 cm de diámetro aproximado, con tapas adecuadas.
5(b).2.5 Arena de cuarzo o arena de mar que pase a través de un tamiz de diez aberturas por cm, pero que no pase a través de un tamiz de 40 aberturas por cm y, si es necesario, lavada con ácido clorhídrico concentrado, después con agua v finalmente secada y calcinada.
Para comprobar si la arena es adecuada, desecar una pequeña cantidad a 98-100 °C hasta peso constante, añadir agua destilada, desecar de nuevo y pesar. No debe haber diferencia entre las dos pesadas.
5(b).2.6 Varillas cortas de vidrio.
5(b).2.7 Baño de agua.
5(b).3 Procedimiento.
5(b).3.1 Preparación de la muestra.‒Si se observa una separación parcial más o menos importante de algunos componentes, por ejemplo, materias proteicas, materia grasa, sales de calcio o lactosa, es necesario mezclar la muestra.
5(b).3.1.1 Leche concentrada o evaporada.‒Abrir el envase al borde de la tapa, verter la leche despacio en otro recipiente y mezclar bien por sucesivos trasvases. La leche o la grasa que se adhiere a la tapa debe reincorporarse a la muestra. Calentar entonces la muestra tapada a una temperatura de 40 °C y mezclar íntimamente removiendo. Dejar enfriar.
5(b).3.1.2 Leche condensada.‒Abrir el bote al borde de la tapa. La leche o grasa adherida a la tapa deberá incorporarse a la muestra. Calentar a 40 °C y mezclar íntimamente removiendo de arriba abajo con una cuchara. Dejar enfriar.
5(b).3.2 Determinación.‒Colocar en la cápsula, aproximadamente, 25 g de arena y una pequeña varilla de vidrio. Secar la cápsula y su contenido, con la tapa abierta, a 98-100 °C durante 2 horas. Colocar la cápsula tapada en un desecador, dejarla enfriar a la temperatura ambiente y pesar. Arrastrar la arena hacia un borde de la cápsula, pesar exactamente y poner en el espacio libre, aproximadamente, 1,5 g de la muestra. Añadir cinco ml de agua destilada, mezclar los líquidos y la arena mediante una varilla de vidrio. Dejar la varilla en la mezcla. Colocar la cápsula en baño de agua hirviendo durante 20 minutos y remover con cuidado la mezcla de cuando en cuando. Colocar la cápsula con la varilla y la tapa en una estufa de desecación a 98°-100 °C durante 1 hora 30 minutos. La tapa se debe colocar al lado de la cápsula. Cubrir la cápsula con la tapa y ponerla en el desecador; dejarla enfriar y pesar. Colocar 1 hora más en la estufa de desecación; dejarla enfriar y pesar como antes. Repetir la desecación hasta que la diferencia en peso entre dos pesadas sucesivas no exceda de 0,5 mg.
5(b).4 Cálculo.
P' = peso en gramos de la muestra después de desecar.
P = peso en gramos de la muestra antes de desecar.
La diferencia entre dos determinaciones repetidas no debe ser mayor de 0,1 por 100 del extracto seco.
5(b).5 Referencia.
1. Norma Internacional FIL-IDF 15: 1961.
6. CENIZAS
6.1 Principio.
Se entiende por contenido en cenizas de la leche el producto resultante de la incineración del extracto seco, expresado en porcentaje en peso, obtenido según el procedimiento descrito a continuación.
6.2 Material y aparatos.
6.2.1 Balanza de sensibilidad: 0,1 mg como mínimo.
6.2.2 Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
6.2.3 Estufa de desecación, regulada a 120 °C.
6.2.4 Horno eléctrico con circulación de aire, provisto de un regulador de temperatura.
6.2.5 Cápsula de platino o de un material inalterable en las condiciones del ensayo de aproximadamente 55 ml de diámetro y 25 de altura.
6.2.6 Baño de agua a temperatura de ebullición.
6.3 Procedimiento.
Colocar la cápsula en la estufa de desecación a 102° ± 2 °C durante 30 minutos. Pasarla luego al desecador, dejarla enfriar a la temperatura ambiente y pesar. Pesar exactamente alrededor de 10 g de leche en la cápsula. Poner la cápsula en baño de agua hirviendo hasta secado por evaporación (aproximadamente siete horas). Incinerar extracto seco, procedente de la desecación anterior, por calentamiento durante 2 o 3 horas en un horno regulado entre 520 y 550 °C (no deben existir en este último partículas carbonosas). Poner a enfriar la cápsula en un desecador. Pesar con una aproximación de 0,5 mg.
6.4 Cálculo.
El contenido en cenizas de la leche, expresado en porcentaje en peso, es igual a

M = peso de la cápsula y de las cenizas después de la incineración y enfriamiento posterior.
m = peso de la cápsula vacía.
P = peso en gramos de la leche empleada en la determinación de las cenizas.
6.5 Observaciones.
El peso de las cenizas es variable según las condiciones de incineración. La técnica descrita anteriormente proporciona los resultados más constantes; las diferencias no pasan generalmente de un 2 por 100 por término medio, y el 95 por 100 por lo menos de los cloruros se encuentran en las cenizas.
Cuando la temperatura del horno se haya elevado ligeramente por encima de 550 °C, determinar los cloruros y corregir en consecuencia el peso de las cenizas.
Si a la muestra se le ha añadido dicromato potásico, es necesario efectuar dos correcciones para obtener un valor aproximado de las cenizas de la leche inicial, operando como sigue:
1.° Determinar el dicromato potásico y restar su peso del de las cenizas.
2.° Determinar los cloruros en las cenizas, expresarlos en ClNa y añadir al peso le las cenizas la diferencia entre los cloruros totales de la leche y los cloruros restantes.
7. DICROMATO POTÁSICO
7.1 Principio.
La determinación se realiza sobre las cenizas de la leche, por reducción de los cromatos mediante una solución de sulfato ferroamónico (sal de Mohr) (SO4)2 Fe (NH4)2 ∙ 6H2O, cuyo exceso se valora con permanganato potásico.
7.2 Reactivos.
7.2.1 Solución acuosa de sal de Mohr de 8 g por 1, para valorar en el momento de su empleo (esta solución se estabiliza por adición de 12 ml de ácido sulfúrico por litro).
7.2.3 Acido sulfúrico (d = 1,84).
7.3 Procedimiento.
Agotar las cenizas por lavados sucesivos con agua destilada; después, con una solución acuosa de ácido sulfúrico al 5 por 100 en volumen, obtener un total de 25 a 30 ml de líquido. Recogerlo en un vaso para valorar. Añadir alrededor de 5 ml de ácido sulfúrico y 20 ml exactamente medidos de la solución sulfúrica de sal de Mohr con la solución valorada de permanganato potásico hasta coloración rosa permanente.
Por otra parte, valorar en las mismas condiciones 20 ml de la misma solución sulfúrica de sal de Mohr mediante la solución valorada de permanganato potásico.
7.4 Cálculo.
El contenido de la leche en dicromato potásico, expresado en porcentaje en peso, es igual a

V = volumen en ml de permanganato potásico empleados en la valoración del exceso de sal de Mohr.
V' = volumen en ml de permanganato potásico empleados en la prueba en blanco.
P = peso en gramos de la leche empleada en la determinación de las cenizas.
8(a). ACIDEZ
(Leches natural, certificada, higienizada y esterilizada)
8(a).1 Principio.
Se entiende por acidez en las leches natural, certificada, higienizada y esterilizada el contenido aparente en ácidos, expresado en g de ácido láctico por 100 ml de leche, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE 34.100 del Instituto de Racionalización del Trabajo.
Un determinado volumen de leche se valora en solución de sosa, empleando solución alcohólica de fenolftaleína y luego se expresa el resultado en peso de ácido láctico, mediante la correspondiente transformación.
8(a).2 Material y aparatos.
Bureta graduada cada 0,05 ml, o cada 0,1 ml que permita apreciar la semidivisión.
8(a).3 Reactivos.
8(a).3.1 Solución 0,111 N (N/9) ó 0,1 N (N/10) de hidróxido sódico (NaOH).
8(a).3.2 Indicador: 0,5 ml de una solución alcohólica de fenolftaleína al 1 por 100.
8(a).4 Procedimiento.
8(a).4.1 Preparación de la muestra.‒Antes del análisis poner la muestra a 20 ± 2 °C y mezclarla cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentar lentamente la muestra a 40 °C, mezclar nuevamente y enfriarla de nuevo a 20 ± 2°C.
8(a)4.2 Determinación.‒Se determina volumétricamente operando sobre 10 ml de leche con solución 0,111 N (N/9) de sosa (NaOH) o sobre 9 ml de leche con solución 0,1 N (N/10). Como indicador se emplean 3,5 ml de solución alcohólica de fenolftaleína al 1 por 100 (dar por terminada la valoración cuando aparece una coloración rosa fácilmente perceptible por comparación con un testigo tomado de la misma leche. Dicha coloración desaparece progresivamente, pero se considera obtenido el viraje cuando el tinte rosa persiste durante unos segundos).
8(a).5 Expresión de los resultados.
Los resultados se expresan en peso de ácido láctico por 100 mililitros de leche, dividiendo por 10 el número de mililitros empleados de solución de sosa.
Si a la muestra de leche se le ha adicionado dicromato potásico, es preciso tener en cuenta la acidez debida a dicho conservador. En el caso de leches no alteradas se puede considerar que 1 g de dicromato potásico aumenta la acidez en las mismas proporciones que 0,6 g de ácido láctico.
8(a).6 Referencia.
1. Instituto Nacional de Racionalización del Trabajo. Una norma española 34.100.
8(b). ACIDEZ
(Leche en polvo)
8(b).1 Principio.
Se entiende por acidez en la leche en polvo el contenido aparente en ácidos expresado en gramos de ácido láctico por 100 g de leche en polvo determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE-34.101 del Instituto Nacional de Racionalización del Trabajo.
El método es aplicable a las leches en polvo, entera y desnatada.
Una determinada cantidad pesada de leche en polvo disuelta en agua se valora con una solución de sosa empleando fenolftaleína como indicador hasta igualarla en color con otra cantidad igual de leche en polvo disuelta y mezclada con acetato de rosanilina.
8(b).2 Material y aparatos.
8(b).2.1 Cápsulas de porcelana blanca de aproximadamente 100 ml de capacidad.
8(b).3 Reactivos.
8(b).3.1 Solución 0,111 N (N/9) ó 0,1 N (N/10) de hidróxido sódico (NaOH) exento de carbonato.
8(b).3.2 Solución indicadora de fenolftaleína: Se disuelve un gramo de fenolftaleína en 110 ml de alcohol etílico (95 a 96 por ciento en volumen), se añade solución N/9 ó N/10 de hidróxido sódico, hasta que una gota dé una débil coloración rosa, y se completa hasta 200 ml con agua destilada.
8(b).3.3 Solución concentrada de acetato de rosanilina: Se disuelven 0,12 g de acetato de rosanilina en 50 ml de alcohol etílico (95 a 96 por 100 en volumen) que contenga 0,5 ml de ácido glacial y se completa hasta 100 ml con alcohol etílico.
8(b).3.4 Solución diluida de acetato de rosanilina: Se diluye un mililitro de solución concentrada de acetato de rosanilina hasta 500 ml con una mezcla, a partes iguales en volumen, de agua destilada y alcohol etílico (95 a 96 por 100 en volumen).
Las soluciones de acetato de rosanilina se conservan en la oscuridad en frascos herméticamente cerrados con tapones de caucho.
8(b).4 Procedimiento.
Se toman dos cápsulas de porcelana blanca de una capacidad aproximada de 100 ml, pesándose en cada una de ellas 1 g de leche en polvo. A una y otra cápsulas se añaden 10 ml de agua hirviente, se agita con la extremidad aplastada de una varilla de vidrio hasta obtener un líquido perfectamente homogéneo y se deja enfriar durante 10 minutos. A una de las cápsulas se le añade 1 ml de solución diluida de acetato de rosanilina y se mezcla. A la otra cápsula se le agrega 1 ml de solución de fenolftaleína y, gota a gota y agitando enérgicamente todo el tiempo, solución valorada de hidróxido sódico hasta obtener una coloración igual a la primera. El tiempo empleado en la valoración no ha de exceder de 20 segundos y ésta se ha de efectuar con una luz difusa.
8(b).5 Cálculo.
El número de mililitros consumidos de solución 0,111 N (N/9) representa la acidez, expresada en gramos, de ácido láctico por 100 g de leche en polvo.
Si se opera con solución 0,1 N (N/10), el número de mililitros multiplicado por 0,9 da el mismo porcentaje de acidez.
8(b).6 Referencia.
1. Instituto Nacional de Racionalización del Trabajo. Una norma española. 34.101.
9. SACAROSA
(Determinación polarimétrica en la leche condensada)
9.1 Principio.
Se entiende por contenido en sacarosa de la leche condensada el contenido en sacarosa no transformada, expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-35: 1966 de la Federación Internacional de Lechería
Este método es aplicable a la leche condensada, entera o desnatada, de composición normal, preparada a partir de leche y sacarosa únicamente que no contenga sacarosa transformada.
El método se basa en el principio de inversión de Clerget: Un tratamiento suave con un ácido hidroliza completamente la sacarosa. La lactosa y los otros azúcares prácticamente no se hidrolizan. La cantidad de sacarosa se deduce del cambio del poder rotatorio de la solución.
Se prepara un filtrado límpido de la muestra, sin mutarrotación debida la lactosa, por tratamiento de la solución con amoníaco seguido de neutralización y clarificación por adiciones sucesivas de soluciones de acetato de cinc y de ferrocianuro potásico,
En una parte del filtrado de sacarosa se hidroliza en las condiciones especiales que corresponden a este tipo de operación.
Partiendo de los poderes rotatorios del filtrado, antes y después de la inversión, se calcula la cantidad de sacarosa.
9.2 Material y aparatos.
9.2.1 Balanza analítica de sensibilidad: 10 como mínimo.
9.2.2 Vasos de precipitados de 100 ml en vidrio.
9.2.3 Matraces graduados, de 200 y 50 ml.
9.2.4 Pipetas de 40 ml.
9.2.5 Probetas graduadas de 25 ml.
9.2.6 Pipetas graduadas de 10 ml.
9.2.7 Embudos filtrantes de diámetro entre 8 y 10 cm y filtros (plegados) de 15 cm de diámetro.
9.2.8 Tubo de polarímetro de 2 cm de longitud, exactamente calibrado.
9.2.9 Polarímetro o sacarímetro.
9.2.9.1 Polarímetro con luz de sodio o con luz verde de mercurio (lámpara de vapor de mercurio con prisma o pantalla Wratten número 77 A), permitiendo lecturas con una precisión por lo menos igual a 0,05 grados de ángulo.
9.2.9.2 Sacarímetro con escala internacional de azúcar utilizando luz blanca que pasa a través de un filtro de 15 ml de una solución al 6 por 100 de dicromato potásico, o bien luz de sodio, y permitiendo la lectura con una precisión por lo menos igual a 0,1 grados de la escala sacarimétrica internacional.
9.2.10 Baño de agua a 60 ± 1 ºC.
9.3 Reactivos.
9.3.1 Solución de acetato de cinc, 2,0 N: Disolver 21,9 g de acetato de cinc cristalizado (C2H3O2)2 Zn ⋅ 2H2O y 3 ml de ácido acético glacial en agua destilada y completar hasta 100 ml,
9.3.2 Solución de ferrocianuro potásico, 1,0 N: Disolver 10,6 gramos de ferrocianuro potásico cristalizado [Fe (CN)6] K4 ⋅ 3H2O en agua destilada y completar hasta 100 ml.
9.3.3 Solución de ácido clorhídrico, 6,35 ± 0,20 N (20-22 por 100).
9.3.4 Solución diluida de amoníaco, 2,0 ± 0,2 N (3,5 por 100).
9.3.5 Solución diluida de ácido acético, 2,0 ± 0,2 N (12 por 100).
9.4 Procedimiento.
9.4.1. Preparación de la muestra.‒Para muestras de productos recientemente preparados en los que no se pueda prever separación alguna apreciable de los componentes. Abrir el recipiente, introducir en él el producto adherido a la tapa y mediante movimientos de arriba abajo, con ayuda de una cuchara, conseguir que se mezclen íntimamente las capas superiores así como el contenido del fondo del recipiente. Trasvasar el contenido de un bote a un frasco provisto de tapón bien adaptado.
Para muestras de productos más antiguos y muestras en las que se pueda prever una separación de componentes, calentar en baño de agua, aproximadamente a 40 °C, hasta que la muestra casi haya alcanzado esta temperatura, abrir el recipiente y operar de la misma manera que arriba. En el caso de un bote, trasvasar el contenido a un frasco, raspar el producto que se haya adherido a las paredes y continuar la mezcla hasta que toda la masa sea homogénea. Cerrar el frasco con una tapadera que se adapte perfectamente. Dejar enfriar.
9.4.2 Comprobación del método.‒Proceder como en 9.4.3, utilizando una mezcla de 100 g de leche o 110 g de leche desnatada y de 18 g de sacarosa pura, que corresponde a 40 g de una leche concentrada conteniendo el 45 por 100 de sacarosa. Calcular la cantidad de sacarosa como en 9.5.1, utilizando en la fórmula I), para W, F y P, la cantidad de leche pesada y la riqueza en materia grasa y proteínas de esta leche, y en la fórmula II), para W, la cifra de 40. La media de los valores encontrados no debe diferir de dicho valor (45 por 100) en más del 0,1 por 100.
9.4.3 Determinación.‒En un vaso de 100 ml pesar aproximadamente 40 g de la muestra bien mezclada con una aproximación de 10 mg, añadir 50 ml de agua destilada caliente (80-90 °C) y mezclar cuidadosamente. Trasvasar cuantitativamente la mezcla a un matraz aforado de 200 ml, enjuagar el vaso con cantidades sucesivas de agua destilada a 60 °C hasta que el volumen total sea de 120 a 150 ml. Mezclar y enfriar a temperatura ambiente. Añadir 5 ml de la solución de amoníaco diluida. Mezclar de nuevo y dejar reposar durante 15 minutos. Neutralizar el amoníaco añadiendo una cantidad equivalente de la solución diluida de ácido acético. Determinar previamente la cantidad exacta de mililitros por valoración de la solución de amoníaco diluida empleando el azul de bromotimol como indicador. Mezclar. Añadir, mezclando suavemente por rotación del matraz inclinado, 12,5 ml de solución de acetato de cinc. De la misma forma que para la solución de acetato, añadir 12,5 ml de solución de ferrocianuro potásico. Poner el contenido del matraz a 20 °C y añadir agua destilada (a 20 °C) hasta alcanzar el enrase de 200 ml.
Hasta este momento todas las adiciones de agua o reactivos deberán haberse efectuado de tal manera que se haya evitado la formación de burbujas de aire y por esa misma razón todas las mezclas se habrán realizado por rotación de matraz y no por agitación violenta. Si se observa la presencia de burbujas de aire antes de alcanzar los 200 ml se pueden eliminar aplicando al matraz una bomba de vacío e imprimiéndole un movimiento de rotación. Tapar el matraz con un tapón seco y mezclar íntimamente sacudiendo con energía. Dejar reposar durante algunos minutos; filtrar a continuación por un papel filtro seco. Despreciar los primeros 25 ml del filtrado.
9.4.3.1 Polarización directa.‒Determinar la rotación óptica del filtrado a 20 ± 2 °C.
9.4.3.2 Inversión.‒Introducir con la pipeta en un matraz graduado de 50 ml, 40 ml del filtrado obtenido de la manera indicada antes. Añadir 8 ml de ácido clorhídrico 6,35 N. Poner el matraz en baño de agua a 60 °C durante 15 minutos, sumergiéndolo hasta el nacimiento del cuello. Mezclar por rotación durante los 5 primeros minutos, al final de los cuales el contenido deberá haber alcanzado la temperatura del baño. Enfriar a 20 °C y completar hasta 50 ml con agua destilada a 20 °C, mezclar y dejar reposar 1 hora a esta temperatura.
9.5 Cálculo.
9.5.1 Riqueza en sacarosa.
I) 
II) 
S = cantidad de sacarosa.
W = peso de la muestra expresado en gramos.
P = porcentaje de proteínas (N × 6,38) de la muestra.
F = porcentaje de materia grasa de la muestra.
V = volumen en mililitros de la muestra diluida antes de filtrar.
D = lectura polarimétrica directa (polarización antes de la inversión).
I = lectura polarimétrica después de la inversión.
L = longitud en decímetros del tubo del polarímetro.
Q = factor de inversión cuyos valores se indican más adelante.
Pesando exactamente 40 g de leche condensada y utilizando un polarímetro con luz de sodio provisto de escala en grados de ángulo y un tubo de 2 cm de longitud, el contenido en sacarosa de las leches condensadas normales (C = 9) a 20° ± 0,1 °C se puede calcular con ayuda de la siguiente fórmula:
S = (D-5/4 I) (2,833 ‒ 0,00612 ⋅ F ‒ 0,00878 P)
Si la medida de la polarización después de la inversión se efectúa a una temperatura diferente de 20 °C, las cifras obtenidas se deben multiplicar por
[I + 0,0037 (T ‒ 20)]
9.5.2 Calores del factor de inversión Q.‒Las fórmulas siguientes dan valores precisos de Q para diversas clases de luz con correcciones, si es necesario, para la concentración y la temperatura.
Luz de sodio y polarímetro con escala en grados de ángulo:
Q = 0,8825 + 0,0006 (C ‒ 9) ‒ 0,0033 (T ‒ 20)
Luz verde de mercurio y polarímetro con escala en grados de ángulo:
Q = 1,0392 + 0,0007 (C ‒9) ‒ 0,0039 (T ‒ 20)
Luz blanca con filtro de dicromato y sacarímetro con escala sacarimétrica:
Q = 2,549 +0,0017 (C ‒ 9) ‒ 0,0095 (T ‒ 20)
En las fórmulas precedentes:
C = porcentaje de azúcares totales en la solución invertida, según la lectura polarimétrica.
T = temperatura de la solución invertida en la lectura del polarímetro.
El porcentaje de azúcares totales C en la solución invertida se puede calcular a partir de la lectura directa y de la variación después de la inversión según el método habitual, utilizando los valores usuales de rotación específica de sacarosa, lactosa y de sacarosa invertida. La corrección 0,0006 (C-9), etcétera, no es exacta más que cuando C es aproximadamente 9; para leche concentrada normal esta corrección se puede despreciar, por ser C próximo a 9.
Variaciones en la temperatura de 20 °C influyen escasamente en la lectura de la polarización directa. Por el contrario, diferencias de más de 0,2 °C, en la lectura de polarización después de la inversión necesitan una corrección. La corrección ‒ 0,003 (T-20), etc., no es exacta más que para temperaturas comprendidas entre 18 °C y 22 °C.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista no debe ser mayor de 0,3 g de sacarosa para 100 g de leche condensada.
9.6 Referencia.
1. Norma Internacional FIL-IDF 35: 1966.
10. HUMEDAD
(Leche en polvo)
10.1 Principio.
Se entiende por humedad de la leche en polvo el contenido en agua libre, es decir, la pérdida de peso, expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-26: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches en polvo, entera y desnatada.
El agua, contenida en la leche en polvo, se elimina por calentamiento de la muestra en una estufa de desecación, a una temperatura de 102° ± 2 °C, hasta peso constante.
10.2 Material y aparatos.
10.2.1 Balanza analítica, de sensibilidad 0,1 mg como mínimo.
10.2.2 Cápsulas apropiadas, preferentemente en aluminio, níquel, acero inoxidable o vidrio. Las cápsulas deberán estar provistas de tapas que se adapten convenientemente, pero que se puedan quitar con facilidad. Las dimensiones más convenientes son: diámetro aproximado, 50 mm; profundidad aproximada, 25 mm.
10.2.3 Un desecador provisto de gel de sílice con indicador de humedad.
10.2.4 Una estufa de desecación bien ventilada, provista de termostato y regulada a 102° ± 2 °C. Es importante que la temperatura sea uniforme en toda la estufa.
10.2.5 Frascos provistos de tapones herméticos para el mezclado de la leche en polvo.
10.3 Procedimiento.
10.3.1 Preparación de la muestra.‒Transvasar toda la muestra de la leche en polvo a un frasco seco y tapado, de un volumen igual al doble, aproximadamente, del de la muestra, mezclar íntimamente por rotación y agitación (en el caso de que no se pueda obtener una homogeneización completa por este procedimiento, extraer, con objeto de realizar determinaciones paralelas, dos muestras en dos puntos, lo más distantes posible el uno del otro).
10.3.2 Determinación.‒Colocar la cápsula destapada y la correspondiente tapa en la estufa de desecación durante 1 hora, a la temperatura de 102° ± 2 °C. Colocar la tapa sobre la cápsula y pasarla de la estufa al desecador. A continuación enfriar a la temperatura ambiente y pesar. Introducir aproximadamente 1 g de leche en polvo en la cápsula, tapar la cápsula y pesar rápidamente con la mayor exactitud posible. Destapar la cápsula y colocarla con su tapa en la estufa a una temperatura de 102° ± 2 °C durante 2 horas. Volver a colocar la tapa, poner la cápsula en el desecador, dejar enfriar a temperatura ambiente y pesar rápidamente con la mayor exactitud posible.
Calentar la cápsula abierta y su tapa a 102° ± 2°C en la estufa durante 1 hora suplementaria, volver a tapar y dejar enfriar en el desecador a temperatura ambiente; pesar de nuevo. Repetir la operación hasta que las pesadas sucesivas no difieran en más de 0,0005 g. La desecación se acaba aproximadamente en 2 horas.
10.4 Cálculo.
Calcular la humedad mediante la fórmula siguiente:

M1 = la masa inicial, en gramos, de la cápsula y su tapa más la leche en polvo utilizada para el análisis.
M2 = la masa final, en gramos, de la cápsula y su tapa más la leche en polvo.
M3 = la masa, en gramos, de la leche en polvo utilizada para el análisis.
La diferencia entre dos determinaciones repetidas no debe sobrepasar el 0,06 por 100 de agua.
10.5 Referencia.
1. Norma Internacional FIL-IDF 26: 1964.
11. ÍNDICE DE SOLUBILIDAD
(Leche en polvo)
11.1 Principio.
Se entiende por índice de solubilidad de la leche en polvo la cantidad de sedimento, expresada en volumen, determinada por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE-34.101 del Instituto Nacional de Racionalización del Trabajo.
Este método es aplicable a la leche en polvo, entera o desnatada.
11.2 Material y aparatos.
11.2.1 Centrífuga con soportes para la colocación de los tubos centrífugos cónicos. La velocidad requerida varía, como a continuación se indica, con el diámetro.
| Diámetro | Revoluciones por minuto | Diámetro | Revoluciones por minuto |
|---|---|---|---|
| 250 milímetros | 1.083 | 450 milímetros | 807 |
| 300 milímetros | 988 | 500 milímetros | 766 |
| 350 milímetros | 916 | 550 milímetros | 730 |
| 400 milímetros | 856 | 600 milímetros | 700 |
El «diámetro» es la distancia entre los fondos internos de dos soportes opuestos, medida a través del centro de rotación de la cabeza de la centrífuga, estando los soportes extendidos horizontalmente.
11.2.2 Tubos de centrífuga cónicos, graduados como se indica a continuación:
De 0 a 1,0 ml en divisiones de 0,1 ml.
De 1,0 a 2,0 ml en divisiones de 0,2 ml.
De 2,0 a 10,0 ml en divisiones de 0,5 ml.
De 10,0 a 20,0 ml en divisiones de 1,0 ml.
y a 50 ml una marca que quede, por lo menos, a 13 mm del borde del tubo.
11.2.3 Mezclador eléctrico.
11.2.4 Tubo sifón de vidrio.
11.3 Procedimiento.
Se añaden 10 ó 13 g de la muestra, según se trate de leche desnatada o completa, respectivamente, a 100 ml de agua destilada, a la temperatura de 24 °C, en el vaso especial del mezclador que seguidamente se pone en éste para agitar durante 90 segundos exactos. Si hay necesidad inmediata de volver a emplear el vaso del mezclador, puede verterse la solución total en un vaso de precipitación apropiado. Se deja reposar la solución hasta que la espuma se separe lo suficiente, para poder quitarla completamente con una cuchara. El período de reposo después de la mezcla no ha de exceder de 15 minutos.
Después de separada la espuma se mezcla completamente con una cuchara durante 5 segundos y, seguidamente, se llena un tubo cónico hasta la marca de 50 ml. Se centrifuga durante 5 minutos a las revoluciones por minuto requeridas, según la tabla del apartado 11 2.1. A continuación se sifona el líquido que sobrenada, hasta 2 ml del nivel del sedimento, teniendo cuidado de no remover éste. Se vierten en el tubo 25 ml de agua destilada a 24 °C y se agita para dispersar el sedimento, desalojándole si fuera necesario con un alambre. Se llena con agua destilada, 24 °C, hasta la marca de 50 ml, se invierte y se agita para la perfecta mezcla del contenido, y se centrifuga de nuevo durante 5 minutos a la velocidad requerida. Se mantiene el tubo en posición vertical, de modo que el nivel superior del sedimento quede a nivel del ojo; se refieren los mililitros de sedimento a la división de la escala más próxima. La lectura se efectúa fácilmente, cuando se observa el tubo, colocado delante de una luz fuerte.
Cuando se opere con leches en polvo parcialmente desnatadas, las cantidades que se han de añadir a los 100 ml de agua destilada son, según sus porcentajes grasos, las siguientes:
Hasta el 4 %: 10,0 gramos.
Desde el 4,1 al 9 %: 10,5 gramos.
Desde el 9,1 al 13 %: 11,0 gramos.
Desde el 13,1 al 17 %: 11,5 gramos.
Desde el 17,1 al 21 %: 12,0 gramos.
Desde el 21,1 al 24 %: 12,5 gramos.
Más del 24 %:13,0 gramos.
11.4 Referencia.
1. Instituto Nacional de Racionalización del Trabajo. Una norma española. 34.101
12. CALCIO
12.1 Principio.
Se entiende por contenido en calcio de la leche la cantidad total de calcio expresada en porcentaje en peso, determinada por el procedimiento expuesto a continuación, que corresponde a la norma FIL-36: 1966 de la Federación Internacional de Lechería.
Este método se aplica a todas las leches líquidas normales, así como a las leches reconstituidas por dilución o disolución de leches concentradas o de leches desecadas.
El calcio total se lleva a disolución por precipitación de las materias proteicas con ácido tricloroacético. El calcio contenido en el filtrado es precipitado bajo forma de oxalato cálcico, que se separa por centrifugación y se valora con permanganato potásico.
12.2 Material y aparatos.
12.2.1 Balanza analítica.
12.2.2 Matraz aforado de 50 ml.
12.2.3 Pipeta para leche de 20 ml.
12.2.4 Papel filtro sin cenizas para filtración lenta.
12.2.5 Centrífuga que pueda desarrollar una aceleración centrífuga de 1.400 g (g = aceleración de la gravedad).
12.2.6 Tubos de centrífuga cilíndricos con fondo redondo de 30 ml, aproximadamente, marcados a 20 ml.
12.2.7 Pipetas de 2 y 5 ml.
12.2.8 Dispositivo de sifón por succión, provisto de un tubo capilar.
12.2.9 Baño de agua, a temperatura de ebullición.
12.2.10 Bureta graduada en 1/50 mm.
12.3 Reactivos.
12.3.1 Ácido tricloroacético: solución acuosa al 20 por 100 (en peso por volumen).
12.3.2 Ácido tricloroacético: solución acuosa al 12 por 100 (en peso por volumen).
12.3.3 Oxalato amónico: solución acuosa saturada en frío.
12.3.4 Rojo de metilo: solución al 0,05 por 100 (en peso por volumen) en alcohol etílico del 96 por 100 (en volumen por volumen).
12.3.5 Ácido acético: solución acuosa al 20 por 100 (en volumen por volumen).
12.3.6 Amoníaco: solución acuosa obtenida mezclando volúmenes iguales de amoniaco al 25 por 100 (en peso por peso) y de agua destilada.
12.3.7 Amoníaco: solución acuosa obtenida diluyendo 2 mililitros de amoníaco al 25 por 100 (en peso por peso) hasta 100 ml, con agua destilada.
12.3.8 Ácido sulfúrico: solución acuosa obtenida añadiendo 20 ml de ácido sulfúrico del 98 por 100 (en peso por peso) a 80 ml de agua destilada.
12.3.9 Permanganato potásico: solución acuosa titulada, 0,02 N.
Todos los reactivos deben ser puros para análisis.
12.4 Procedimiento.
12.4.1 Preparación de la muestra.‒Antes del análisis, poner la muestra a 20° ± C y mezclar con cuidado. Si no se obtiene una dispersión homogénea de la materia grasa, calentar lentamente la muestra a 40 °C, mezclar suavemente y enfriar a 20° + 2 °C.
12.4.2 Defecación de la muestra.‒En un matraz aforado de 50 ml pesar exactamente alrededor de 20 g de leche, con una aproximación de 10 mg. Añadir poco a poco mientras se agita una solución acuosa de ácido tricloroacético al 20 por 100 y completar hasta 50 ml con este reactivo. Agitar fuertemente durante algunos segundos. Dejar reposar 30 minutos. Filtrar sobre papel filtro sin cenizas. El filtrado obtenido debe ser límpido.
12.4.3 Precipitación del calcio en forma de oxalato y separación del oxalato.‒En un tubo de centrífuga cilíndrico de fondo redondo, introducir 5 ml del filtrado límpido; después 5 mililitros de ácido tricloroacético al 12 por 100, 2 ml de una solución acuosa saturada de oxalato de amonio, dos gotas de solución alcohólica de rojo de metilo y 2 ml de ácido acético al 20 por 100. Mezclar mediante agitación circular y añadir poco a poco solución de amoníaco (12.3.6) hasta coloración amarillo pálido; después, algunas gotas de ácido acético al 20 por 100 hasta coloración rosa. Dejar reposar 4 horas a la temperatura ambiente.
Diluir hasta 20 ml con agua y centrifugar 10 minutos a 1.400 g (g = aceleración de la gravedad). Mediante un dispositivo de succión, decantar el líquido límpido que sobrenada. Lavar las paredes del tubo de centrifugación (sin remover el depósito de oxalato cálcico) con 5 ml de solución de amoníaco diluido (12.3.7). Centrifugar 5 minutos a 1.400 g. Decantar el líquido que sobrenada con el dispositivo de succión. Proceder a tres lavados sucesivos.
12.4.4 Valoración del oxalato.‒Después de haber extraído con sifón el agua del ultimo lavado, añadir 2 ml de solución acuosa de ácido sulfúrico y 5 ml de agua destilada sobre el precipitado de oxalato de calcio. Poner el tubo en baño de agua hirviendo, y cuando el oxalato esté completamente disuelto, valorar con solución de permanganato potásico 0,02 N, hasta coloración rosa persistente. La temperatura debe permanecer superior a los 60 °C durante la valoración.
12.4.5 Ensayo en blanco.‒Efectuar un ensayo en blanco con todos los reactivos, utilizando 20 ml de agua destilada en vez de leche.
12.5 Cálculo.

V = volumen en ml de MnO4K (0,02 N) gastados en la valoración de la muestra.
v = volumen en ml de MnO4K (0,02 N) gastados en el ensayo blanco.
P = peso en gramos de la muestra inicial (alrededor de 20 g).
K = coeficiente de corrección del volumen del precipitado resultante de la precipitación tricloroacética.
Para leche entera (3,5 a 4,5 por 100 de materia grasa): K = 0,972.
Para leche con 3 por 100 de materia grasa: K = 0,976.
Para leche con 2 por 100 de materia grasa: K = 0,980.
Para leche con 1 por 100 de materia grasa: K = 0,985.
Para leche desnatada: K = 0,989.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista, no debe ser mayor de 0,002 g de calcio por 100 g de leche.
12.6 Referencia.
1. Norma Internacional FIL-IDF 36: 1966.
13. FÓSFORO
13.1 Principio.
Se entiende por contenido en fósforo de la leche la cantidad total de fósforo expresada en porcentaje en peso, determinada por el procedimiento expuesto a continuación, que corresponde a la norma FIL-42: 1967 de la Federación Internacional de Lechería.
Este método se aplica a todas las leches líquidas normales, así como a las leches reconstituidas por dilución o disolución de las leches concentradas o de las leches desecadas.
Las materias orgánicas de la leche se destruyen por mineralización en seco (incineración). El fósforo se determina colorimétricamente, reduciendo el fosfomolibdato de amonio con diaminofenol (amidol) y midiendo la densidad óptica de la solución obtenida.
13.2 Material y aparatos.
13.2.1 Balanza analítica.
13.2.2 Cápsulas de platino o de cuarzo (de 55 mm de diámetro aproximadamente) provista de vidrio de reloj.
13.2.3 Baño de agua.
13.2.4 Pipetas de 1, 2, 5 y 10 ml.
13.2.5 Matraces aforados, de 25 ml con tapones esmerilados, de 100 y de 1.000 ml.
13.2.6 Horno mufla.
13.2.7 Estufa a 105° ± 2 °C.
13.2.8 Espectrofotocolorímetro.
13.3 Reactivos.
13.3.1 Acido clorhídrico (ClH) 1,0 N.
13.3.2 Acido perclórico (ClO4H) al 65 por 100 (densidad, 1,61 gramos por ml a 20 °C).
13.3.3 Solución de amidol: Disolver en agua destilada 1 gramo de amidol (diclorhidrato de 2,4 diaminofenol: (NH2)2 C6H3OH. 2ClH) y 20 g de metabisulfito de sodio o pirosulfito de sodio (S2O Na2). Completar hasta 100 ml en un matraz aforado con tapón esmerilado. Preparar cada día una solución nueva.
13.3.4 Solución de molibdato: Disolver en agua destilada 8,3 g de molibdato de amonio: Mo7O24, (NH4)6 ∙ 4H2O. Completar hasta 100 ml.
13.3.5 Solución acuosa de fosfato monopotásico (a partir de PO4H2K, desecado durante 2 horas a 105 °C), que permite trazar una curva de diferencia para la determinación del fósforo: Pesar 4,393 g de PO4H2K, disolver en agua destilada y completar hasta 1.000 ml (solución A). Diluir 10 ml de solución A hasta 1.000 ml (solución B). Un ml de solución B = 10 microgramos de P.
Todos los reactivos deben ser puros para análisis.
13.4 Procedimiento.
13.4.1 Preparación de la muestra: Antes del análisis poner la muestra a 20 ± 2 °C y mezclar cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentar lentamente la muestra a 40 °C, agitar suavemente y enfriar a 20 ± 2 °C
13.4.2 Ensayo en blanco: Efectuar un ensayo en blanco con todos los reactivos, operando como se prescribe en 13.4.3.3, pero reemplazando los 5 ml de la dilución por 5 ml de agua destilada.
13.4.3 Determinación.
13.4.3.1 Mineralización por vía seca: En una cápsula de platino o de cuarzo pesar exactamente alrededor de 10 g de leche con una aproximación de 10 mg. Evaporar, hasta sequedad, al baño de agua hirviendo. Después de la desecación completa, calcinar en la mufla a una temperatura comprendida entre 500 °C y 550 °C hasta la obtención de cenizas blancas.
13.4.3.2 Preparación de la solución de cenizas: Después de enfriar la cápsula, cubrirla con vidrio de reloj, disolver las cenizas en 2 ó 3 ml de ácido clorhídrico normal y diluir con agua destilada. Transvasar la solución de las cenizas a un matraz aforado de 100 ml, lavar el vidrio de reloj y la cápsula, recogiendo las aguas de lavado en el matraz. Completar hasta 100 ml con agua destilada, agitar y filtrar. Tomar 10 ml del filtrado, introducirlos en un matraz de 100 ml. Completar hasta 100 ml con agua destilada y agitar.
13.4.3.3 Determinación colorimétrica del fósforo: Tomar 5 mililitros de la dilución preparada en 13.4.3.2 e introducirlos en un matraz aforado de 25 ml.
Añadir sucesivamente 2 ml de ácido perclórico, 2 ml de la solución de amidol, 1 ml de la solución de molibdato de amonio. Completar hasta 25 ml con agua destilada y mezclar. Esperar 5 minutos y medir la densidad óptica utilizando cubeta de 1 cm en un espectrofotocolorímetro a 750 nanómetros. Buscar en la curva patrón la cantidad de fósforo contenida en el matraz de 25 ml, expresada en microgramos y correspondiente a la densidad óptica leída.
13.4.3.4 Determinación de la curva patrón: En cuatro matraces aforados de 25 ml introducir 3, 5, 7 y 10 ml de la solución R. Las cantidades así introducidas son iguales a 30, 50, 70 y 100 microgramos de fósforo. Proceder a continuación como en 13.4.3.3. Situar sobre una gráfica las densidades ópticas obtenidas en función de las cantidades de fósforo presentes en los matraces. Trazar la curva patrón (que es una recta).
13.5 Cálculo.
El contenido en fósforo de la leche expresada en g de fósforo por 100 g de leche viene dado por la fórmula siguiente:

m = masa de fósforo, expresada en microgramos, obtenida según 13.4.3.4 y contenida en el matraz de 25 ml con la muestra de aproximadamente 50 mg de leche.
m' = masa de fósforo, expresada en microgramos, obtenida según 13.4.3.4 y contenida en el ensayo en blanco.
M = masa de leche, expresada en gramos, empleada para la mineralización.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista no debe ser mayor de 0,008 g de fósforo por 100 g de leche.
13.6 Referencia.
1. Norma Internacional FIL-IDF 42: 1967.
Mantequilla
1. ÍNDICE DE ACIDEZ DE LA GRASA
1.1 Principio.
El índice de acidez de la materia grasa en la mantequilla es el número de mg de hidróxido de potasio que se necesita para neutralizar 1 g de materia grasa.
La materia grasa, después de separarla por fusión de la mantequilla, se disuelve en una mezcla de alcohol-éter, y luego se titula con una solución alcalina valorada.
1.2 Material y aparatos.
1.2.1 Balanza analítica.
1.2.2 Matraces erlenmeyer de 300 ml.
1.2.3 Bureta graduada contrastada en divisiones de 0,1 ml.
1.3 Reactivos.
Los reactivos que se utilicen deben ser de calidad pura para análisis.
1.3.1 Solución alcohólica de hidróxido de potasio, 0,1 N, valorada. Utilizar alcohol etílico absoluto.
1.3.2 Mezcla de volúmenes iguales de alcohol etílico de 95-96 por 100 (v/v) o de alcohol desnaturalizado con metanol y de éter dietílico, neutro a la fenolftaleína.
1.3.3 Solución neutra de fenolftaleína al 1 por 100 (m/v), en alcohol etílico desnaturalizado con metanol.
Nota: Alcohol desnaturalizado con metanol: 100 alcohol absoluto y 5 metanol.
1.4 Procedimiento.
Para separar la materia grasa, fundir la muestra, dejarla reposar a 50°-60° 2 ó 3 horas, decantar y filtrar con papel de filtro seco. Filtrar nuevamente si el primer filtro no está claro. Utilizar la materia grasa fundida, clarificada, bien mezclada.
En un matraz erlenmeyer pesar con precisión de 1 miligramo 5-10 g de materia grasa. Añadir 50-100 ml de la mezcla de alcohol etílico y éter dietílico y disolver en esta mezcla la materia grasa. Añadir 0,1 ml de la solución de fenolftaleína. Valorar con la solución alcalina hasta que aparezca una coloración rosa pálido que persista al menos 10 segundos.
1.5 Cálculo.

V = volumen, en ml, de la solución alcalina empleada.
t = normalidad de la solución alcalina.
A = masa, en gramos, de la porción ensayada.
La diferencia entre los resultados de dos determinaciones paralelas no debe ser mayor de 0,1 mg de hidróxido de potasio por 1 g de materia grasa.
1.6 Referencia.
1. Norma FIL-IDF - G A - 1-69.
2. ÍNDICE DE REFRACCIÓN DE LA GRASA
2.1. Principio.
El índice de refracción de la materia grasa en la mantequilla es la razón entre la velocidad de una luz de longitud de onda determinada (luz de sodio) en el aire y la velocidad de esta misma luz en la materia grasa de la mantequilla a 40 °C.
Mediante un refractómetro apropiado se determina el índice de refracción de la materia grasa obtenida por fusión de la mantequilla.
2.2 Material y aparatos.
2.2.1 Refractómetro provisto de escala graduada en índices de refracción, que permita efectuar lecturas hasta la tercera cifra decimal y cuyos prismas pueden calentarse mediante un líquido circulante, regulándose la temperatura termostáticamente con una aproximación de ± 0,1 °C.
2.2.2 Luz de sodio. Se puede utilizar también la luz blanca si el refractómetro tiene un dispositivo de compensación cromática.
2.3 Procedimiento.
Para separar la materia grasa, fundir la muestra y dejarla reposar 2 ó 3 horas a 50°-60 °C; decantar y filtrar con papel de filtro seco. Filtrar nuevamente si el primer filtrado no está claro. Utilizar la materia grasa fundida, clarificada, bien mezclada sin agua.
Preparar y regular el refractómetro según el modo de empleo del aparato. Ajustar la temperatura del líquido circulante a 40° ± 0,1 °C.
Colocar algunas gotas de materia grasa, preparada en la forma anteriormente descrita, entre los prismas del refractómetro, de manera que se llene por completo el espacio comprendido entre ellos. Dejar transcurrir algunos minutos para que la materia grasa alcance la temperatura de los prismas. Efectuar la lectura con cuatro cifras decimales. Corregir el índice de refracción obtenido añadiendo 0,000045 unidad de índice de acidez si este último (determinado según 1) es igual o superior a dos. Redondear la cuarta cifra decimal.
2.4 Cálculo.
La diferencia entre los resultados de determinaciones paralelas no debe ser mayor de 0,0002 de la unidad de índice de refracción.
2.5 Referencia.
1. F.A.O. - B-5 - 1967.
3. CLORURO SÓDICO
3.1 Principio.
Se entiende por contenido de sal (cloruro de sodio) de la mantequilla el porcentaje en masa de la sal (cloruro de sodio) determinado por el procedimiento que se describe a continuación.
Después de fundir la mantequilla añadiendo agua hirviendo, los cloruros de las mezclas se valoran con una solución de nitrato de plata, empleando cromato de potasio como indicador, según el procedimiento de Mohr, y se calcula el contenido de sal.
3.2 Material y aparatos.
3.2.1 Balanza analítica.
3.2.2 Matraces erlenmeyer de 250 ml de capacidad.
3.2.3 Bureta graduada y contrastada en divisiones de 0,1 mililitros.
3.3 Reactivos.
3.3.1 Solución valorada de nitrato de plata, 0,1 N.
3.3.2 Solución de cromato de potasio al 5 por 100 (m/v) en agua destilada.
3.4 Procedimiento.
Ablandar la muestra en un recipiente cerrado, calentándola en baño de agua a la temperatura más baja posible, con objeto de no romper la emulsión. Frecuentemente es adecuada una temperatura comprendida entre 23° y 28 °C y en ningún caso la temperatura podrá exceder de 39 °C. Agitar el recipiente que contiene la muestra a intervalos frecuentes durante el proceso de ablandamiento con objeto de que la muestra se mezcle homogéneamente. Sacar el recipiente del baño de agua y agitarlo vigorosamente a intervalos frecuentes hasta que la muestra se haya enfriado adquiriendo una consistencia espesa y cremosa. Esta operación puede realizarse con un agitador mecánico.
Efectuar una determinación en blanco, empleando los mismos reactivos, en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación.
Pesar con una precisión de 10 mg, 5 mg de muestra e introducirla en un matraz erlenmeyer.
Añadir cuidadosamente 100 ml de agua destilada hirviendo. Dejar en reposo durante 5-10 minutos, agitando por rotación de cuando en cuando, mientras se enfría a una temperatura de 50-55 °C (temperatura de valoración). Añadir 2 ml de solución de cromato de potasio. Mezclar agitando por rotación. Mientras se agita continuamente, valorar con la solución de nitrato de plata hasta que el cambio de color anaranjado pardo persista durante 30 segundos.
3.5 Cálculo.
El contenido de sal (expresado en porcentaje por masa de NaCl) es:

t = normalidad de la solución de nitrato de plata.
v1 = volumen, en ml, de solución de nitrato de plata, utilizados en la valoración.
ve = volumen, en ml, de solución de nitrato de plata en el ensayo en blanco.
a = masa, en gramos, de la muestra utilizada.
La diferencia entre los resultados de dos determinaciones paralelas no deberá ser mayor de 0,02 g de cloruro de sodio por 100 g de producto.
3.6 Referencia.
1. Norma FIL-12A - 1969.
4. AGUA, EXTRACTO SECO MAGRO Y GRASA EN UNA SOLA MUESTRA
4.1 Principio.
Se define el contenido de agua en la mantequilla como la pérdida de masa, expresado como porcentaje, en masa, según se determina por el procedimiento descrito.
Se define el contenido de extracto seco magro en la mantequilla como el porcentaje, en masa, de sustancias, según se determina.
Se define el contenido de materia grasa en la mantequilla como el porcentaje, en masa, que se obtiene restando de 100 el contenido de agua y el del extracto seco magro.
El contenido de agua se determina gravimétricamente secando una cantidad conocida de mantequilla a 102° ± 2 °C.
El contenido de extracto seco magro se determina gravimétricamente después de extraer con éter de petróleo la grasa de la mantequilla secada.
4.2 Material y aparatos.
4.2.1 Balanza analítica con una tolerancia de 0,1 mg.
4.2.2 Estufa de desecación bien ventilada y controlada con termostato ajustada para que funcione a una temperatura de 102° ± 2 °C).
4.2.3 Cápsulas metálicas, de porcelana o de vidrio, resistentes a la corrosión, que tengan por lo menos 25 mm de altura y 50 mm de diámetro.
4.2.4 Crisoles filtrantes de vidrio sintetizado (porosidad número 3) con matraces de filtración a la trompa.
4.2.5 Varilla con pieza final de material adecuado.
4.3 Reactivos.
Éter de petróleo con límites de ebullición entre 30 °C y 60 °C. Este reactivo no debe dejar ningún residuo por evaporación.
4.4 Procedimiento.
4.4.1 Preparación de la muestra.‒Excepto cuando el mezclado no se considere necesario, la muestra debe mezclarse agitando con una varilla o con un agitador mecánico, lo más rápidamente posible, sin exceder de 1 minuto. La temperatura del mezclado deberá estar comprendida normalmente entre 23 °C y 28 °C, pero en ningún caso deberá exceder de 35 °C. Antes de pesar, la muestra deberá ponerse siempre a la temperatura ambiente.
4.4.2 Determinación de agua.‒Secar la cápsula en la estufa hasta masa constante. Dejar enfriar la cápsula en desecador hasta la temperatura ambiente (30-35 min.) y pesar. Pesar en la cápsula, de 5 a 10 g de la muestra de mantequilla. Mantener la cápsula en la estufa durante una hora, por lo menos. Dejar que se enfríe la cápsula en desecador a la temperatura ambiente (de 30-35 min.) y pesar. Repetir el secado a intervalos de media hora hasta masa constante (variación igual o inferior a 0,5 miligramos). Todas las pesadas se harán con una precisión de 0,1 miligramo. En el caso de que aumente la masa, se toma la masa mínima para el cálculo. No deben emplearse en esta determinación materiales absorbentes.
4.4.3 Determinación del extracto seco magro.‒Secar el crisol de vidrio filtrante en la estufa hasta masa constante. Dejar enfriar el crisol a temperatura ambiente (30-35 min.) y pesar. Añadir de 10 a 15 ml de éter de petróleo caliente a la cápsula, que contiene el extracto seco procedente de la determinación de agua, de manera que se disuelva la grasa.
Separar la mayor cantidad posible del sedimento adherido a la cápsula utilizando una varilla y pasar cuantitativamente la solución sobre la punta de la varilla al crisol. Repetir las operaciones cinco veces. Lavar el sedimento que queda en el crisol con 25 ml de éter de petróleo caliente. Secar la cápsula y el crisol en la estufa durante 2 horas. Dejar que la cápsula y el crisol se enfríen a la temperatura ambiente (30-35 min.) y pesar. Repetir las operaciones durante períodos de 30 minutos a la temperatura de secado hasta que la masa no disminuya más. Todas las pesadas se harán con una precisión de 0,1 mg.
4.5 Cálculo.
4.5.1 Método de cálculo de contenido de agua.‒Emplear la fórmula:

Siendo:
M = masa, en gramos de la muestra.
n = masa, en gramos, de la muestra después de secar.
4.5.2 Método de cálculo del extracto seco magro.‒Emplear la fórmula

Siendo:
A1 = masa, en gramos, del crisol vacío.
A2 = masa, en gramos, del crisol conteniendo sedimento.
B1 = masa, en gramos, de la cápsula vacía.
B2 = masa, en gramos, de la cápsula con sedimento residual.
M = masa, en gramos, de la muestra.
4.5.3 Método de cálculo del contenido de grasa.
Grasa % = 100 ‒ (E + S)
E = porcentaje, en masa, de agua (calculada en 4.5.1).
S = porcentaje, en masa, de extracto seco magro (calculado en 4.5.2).
Para la determinación del contenido de agua, la diferencia entre el resultado de determinaciones paralelas no deberá exceder de 0,1 g de agua para 100 g de mantequilla.
Para la determinación del contenido de extracto seco magro, la diferencia entre resultados de determinaciones paralelas no deberá exceder de 0,05 g de extracto seco magro para 100 g de mantequilla.
5. DETECCIÓN DE GRASA VEGETAL EN GRASA DE LECHE POR CROMATOGRAFÍA DE GASES DE ESTEROLES
5.1 Principio.
Los digitónidos de esterol se disuelven en una mezcla de formamida y dimetilformamida, y los esteroles liberados se extraen con pentano, y se separan por cromatografía de gases. Si se obtiene en el cromatograma un pico con el tiempo de retención del beta-sitosterol, se concluye la presencia de grasa vegetal en la muestra de grasa examinada.
5.2 Material y aparatos.
5.2.1 Cromatógrafo de gases, equipado de un detector de ionización de llama, con un inyector de plata o de vidrio con un sistema de inyección directa en la columna y con un registrador.
5.2.2 Columna de cromatografía de gases, de vidrio o de acero inoxidable, en forma de U o en espiral, de 1 ó 2 m de longitud, diámetro interior de 3-4 mm Se recomienda el vidrio, pues algunos tipos de acero inoxidable dan resultados erróneos por alteración de los esteroles.
5.2.3 Microjeringa, capaz de proporcionar dosis de 5 ó 10 microlitros.
5.3 Reactivos.
5.3.1 Mezcla de volúmenes iguales de formamida y dimetilformamida.
5.3.2 n-Pentano.
5.3.3 Relleno de la columna: fase estacionaria de goma de silicona (tipo metílico), estable hasta por lo menos 300 °C que impregna en m2 a 4 por 100, una tierra de diatomeas calcinada, lavada al ácido y silanizada, de granulometría 80/100 ó 100/120 de malla.
5.3.4 Solución para el ensayo de sensibilidad: 1 mg de colesterol de 1 ml de n-pentano, recientemente preparado a partir de la grasa de leche, como se describe en 5.4.2.
5.3.5 Solución para el ensayo de resolución de los picos: 0,9 mg de fitosterol de aceite de colza y 0,1 mg de colesterol en 1 ml de n-pentano. Los esteroles deben estar recientemente preparados según el procedimiento descrito en 5.4.2.
5.3.6 Solución para el ensayo de referencia: 1 mg de fitosterol de aceite de soja en 1 ml de n-pentano recientemente preparado, según se describe en 5.4.2.
5.3.7 Gas portador, nitrógeno.
5.3.8 Hidrógeno.
5.3.9 Oxígeno o aire.
5.4 Procedimiento.
5.4.1 Preparación de la muestra.‒Fundir aproximadamente 50 g de la muestra de mantequilla en una estufa corriente a temperatura inferior a 50 °C hasta separación de las fases acuosa y grasa. Separar la capa grasa por decantación y clarificar la grasa en una estufa a una temperatura de aproximadamente 40 °C filtrando sobre un papel de filtro seco y evitando que pase la fase acuosa sobre el filtro.
5.4.2 Preparación de esteroles.‒Pesar en un matraz erlenmeyer de 500 ml, aproximadamente 15 g de materia grasa con una precisión de 0,1 g. Añadir 10 ml de la solución de hidróxido de potasio y 20 ml de etanol (95-96 por 100 v/v). Colocar sobre el matraz erlenmeyer el refrigerante de aire, calentar en baño de agua hirviendo, agitando por rotación, hasta que la solución se haga clara, y continuar la ebullición media hora más.
Añadir 60 ml de agua y luego 180 ml de etanol (95-96 por 100 v/v) y llevar la temperatura a aproximadamente 40 °C. Añadir 30 ml de la solución alcohólica de digitonina (1 por 100), agitar y dejar enfriar. Colocar el matraz en un refrigerante regulado a 5 °C, aproximadamente, durante 12 horas o una noche.
Recoger el precipitado del digitónido de esterol por filtración sobre un papel de filtro de velocidad media en un embudo Buchner (8 cm de diámetro). Lavar el precipitado con agua aproximadamente a 5 °C, hasta que el filtrado no forme espuma, luego lavar una vez con 25-50 ml de etanol (95-96 por 100 v/v) y después una vez con 25-50 ml de éter dietílico.
Desecar el papel de filtro con el precipitado en un vidrio de reloj en estufa a 102 ± 2 °C, durante 10 a 15 minutos. Separar el precipitado en forma de película, plegando en dos partes el papel de filtro.
Disolver en un pequeño tubo de ensayo, aproximadamente, 10 mg de digitónido de esterol en 0,5 ml de una mezcla de volúmenes iguales de formamida y dimetil-formamida. Calentar ligeramente, si es necesario. Después enfriar, añadir 2,5 ml de n-pentano y agitar. Dejar reposar hasta que la separación entre las capas sea neta y usar la capa superior, que contiene los esteroles liberados, para el análisis cromatográfico.
5.4.3 Condiciones de la cromatografía de gases.‒Temperatura de la columna: 220-250 °C. Temperatura del sistema de inyección, si puede calentarse por separado: 20-40 °C por encima de la temperatura de la columna. Gasto de nitrógeno: 30/60 ml/min. Desconectar el detector y equilibrar las columnas nuevas en estas condiciones durante 16-24 horas. Conectar el detector, encender la llama y regular el gasto de hidrógeno y oxigeno o aire para obtener una altura de llama y una sensibilidad adecuada. Poner en marcha el registrador y dejar que el papel se desenrolle a una velocidad adecuada, ajustar el cero y el atenuador. Si la línea de base es estable, el aparato está listo para usarse.
5.4.4 Ensayo de sensibilidad.‒Inyectar 3 a 5 mcl de la solución para el ensayo de sensibilidad (5.3.4). Sólo aparecerá un pico de colesterol en el cromatograma (fig. 5.1). Ajustar el atenuador de modo que se utilice aproximadamente toda la escala del registrador.
5.4.5 Ensayo de resolución de los picos.‒Inyectar 3 a 5 mcl de la solución para el ensayo de resolución de los picos (5.3.5). Aparecerán en el cromatograma los picos de colesterol, brasicosterol, camposterol (fig. 5.II) Medir las distancias de retención (distancia desde el punto de inyección hasta el punto de altura máxima del pico) de los picos, dch para el colesterol, db para el brasicosterol, dc para el camposterol y el ds para el beta-sitosterol y las anchuras de la base de los picos (dimensión de retención entre las intersecciones con la línea de base de las tangentes en los puntos de inflexión situados en la parte anterior y posterior del pico). Wch para el colesterol y Wb para el brasicosterol. La resolución de los picos, expresada por la fórmula
PR = 2(db ‒ dch) (Wb + Wch)
debe ser por lo menos igual a 1.
Calcular los tiempos de retención relativos (colesterol 1,00) para el brasicosterol, el camposterol y el beta-sitosterol.
5.4.6 Ensayo de referencia.‒Inyectar 3 a 5 mcl de la solución para el ensayo de referencia (5.3.6). Los picos de camposterol, estigmasterol y de beta-sitosterol deben aparecer sobre el cromatograma (fig. 5.III). Medir las distancias de retención de los picos, dc para el camposterol, dst para el estigmasterol y ds para el beta-sitosterol.
Calcular los tiempos de retención relativos, que son, aproximadamente:
Colesterol: 1,00 (aproximadamente 15 min.).
Brasicosterol: 1,13-1,15.
Camposterol: 1,32-1,34.
Estigmasterol: 1,44-1,46.
Beta-sitosterol: 1,66-1,68.
5.4.7 Análisis.‒Inyectar 3 a 5 mcl de la solución a analizar y dar al botón del atenuador hasta obtener un factor de atenuación cuatro veces inferior (se obtiene generalmente en dos pases del botón). Registrar el cromatograma.
5.5 Cálculo.
Si en el cromatograma un pico tiene un tiempo de retención relativo igual al de beta-sitosterol y una altura que corresponde al menos a un 2 por 100 de la escala, se concluye la presencia de beta-sitosterol y la muestra de grasa examinada, a partir de la cual se han aislado los esteroles, se considera que contiene grasa vegetal. La presencia en el cromatograma de picos de otros fitosteroles, como el camposterol o el estigmasterol, refuerza esta conclusión.
Por este método se puede demostrar la presencia de al menos un 0,5 por 100 de beta-sitosterol en la mezcla de esteroles. El límite de detección de la grasa vegetal en la grasa de leche no se puede indicar porque depende del contenido en betasitosterol de la grasa añadida, es decir, de la naturaleza de esta grasa o de la mezcla de grasas añadidas a la grasa de leche.
5.6 Referencia.
1. Norma Internacional FIL-1DF 54: 1970.

6. FOSFATASA RESIDUAL EN MANTEQUILLA
6.1 Principio.
El ensayo se basa en la acción del enzima fosfatasa sobre el substrato fenil fosfato disódico, con liberación del fenol y fosfato. La cantidad de fenol liberada se determina por adición de un reactivo que da color azul en presencia de fenol.
Cantidades superiores a dos equivalentes de fenol en 0,5 g de mantequilla indican una pasterización insuficiente.
6.2 Material y aparatos.
6.2.1 Cuchillo o espátula de acero inoxidable.
6.2.2 Baño de agua a 37°-38 °C.
6.2.3 Termómetro.
6.2.4 Pipetas de 1 ml.
6.2.5 Embudo de 5 cm de diámetro.
6.2.6 Papel Whatman núm. 42 o núm. 2.
6.2.7 Tubos graduados a 5 y 10 ml.
6.2.8 Fotómetro con filtro de transmitancia máxima a 610 nm.
6.2.9 Centrífuga
6.2.10 Pera de goma para pipetar.
6.3 Reactivos.
6.3.1 Tampón hidróxido-borato de bario: Disolver 18 g de BA (OH)2BH2O y 8 B de H3BO3 en agua y diluir a 1 litro con agua.
6.3.2 Tampón de desarrollo de color: PH 9,8 ± 0,15 a 25 °C. Disolver 6,0 g de metaborato sódico (NaBO2) y 20 g de NA Cl en agua y diluir a 1 litro con agua.
6.3.3 Tampón de dilución de color: Diluir 100 ml de tampón de desarrollo de color a 1 litro de agua.
6.3.4 Sustrato tampón: Disolver 0,10 g de fenil fosfato disódico cristalino libre de fenol en 100 ml de tampón hidróxido-borato de bario (6.3.1) (los cristales de Na2C6H5PO4 deben guardarse en congelador o en desecador). Si Na2C6H5PO4 no está libre de fenol, purificarlo como sigue: Disolver 0,5 g con 4,5 ml de agua, añadir 0,5 ml de tampón hidróxido-borato de bario (6.3.1) y dos gotas del reactivo BQC (6.3.5) y dejar reposar 30 minutos. Extraer el color con 2,5 ml de alcohol butílico (6.3.7) y dejar reposar hasta que el alcohol se separe. Retirar el alcohol con un cuentagotas y desecharlo. Diluir 1,0 ml de la solución acuosa a 100 ml de tampón hidróxido-borato de bario (6.3.1). Calentar la solución a 85 °C 2 minutos, tapar inmediatamente y conservar en refrigerador. La solución es estable un año si las porciones son recogidas con mínima exposición a la atmósfera.
6.3.5 Solución BQC (s,6-dibromoquinona cloroimida) o reactivo de Gibbs: Disolver 40 mg BQC en polvo en 100 ml de alcohol absoluto o metanol y pasarlo a un frasco cuentagotas oscuro. El reactivo permanece estable por lo menos un mes, si se guarda en congelador; no usarlo después de que empiece a ponerse pardo. Guardar BQC en polvo en congelador o en desecador (nota: ha habido explosiones del reactivo BQC guardado en botellas en la estantería de reactivos). Comprobar los nuevos lotes de BQC antes de usarlo, preparando una curva patrón con fenol y comparando la curva obtenida con la de BQC que se sabe es satisfactorio. Repetir la prueba al menos semestralmente.
6.3.6 Solución de sulfato de cobre para los patrones: Disolver 0,05 g CuSO45H2O en agua y diluir a 100 ml.
6.3.7 Alcohol butílico: Usar alcohol butílico normal, punto de ebullición 116-118 °C. Para ajustar el pH, mezclar 1 litro con 50 ml de tampón de desarrollo de color. Guardar en recipiente con tapón de vidrio.
6.3.8 Solución patrón de fenol:
6.3.8.1 Solución madre: Pesar exactamente 1.000 g de fenol puro, llevarlo a un matraz aforado de 1 litro, diluirlo con agua hasta 1 litro y mezclar (1 ml = 1 mg de fenol). La solución es estable varios meses en refrigerador.
6.3.8.2 Patrones de trabajo: Diluir 10,0 ml de la solución madre con agua hasta 1 litro y mezclar (1 ml = 10/ug 0,00001 g o 10 unidades de fenol). Usar esta solución patrón para preparar soluciones patrón más diluidas; p. e., diluir 5, 10, 30, 50 ml con agua hasta 100 ml para preparar soluciones patrón, que contenga 0,5; 1,0; 3,0 y 5,0 mg o unidades de fenol ml, respectivamente. Guardar estas soluciones patrón en refrigerador no más de una semana.
Análogamente preparar, a partir de la solución madre (6.3.8.1), soluciones patrón que contengan 20, 30 y 40 unidades/ml.
Medir las cantidades adecuadas de las soluciones patrón de trabajo en una serie de tubos (preferiblemente graduados a 5,0 y 10,0 ml) para conseguir un intervalo adecuado de patrones, según se necesite, que contengan O (control o prueba en blanco), 0,5; 1,0; 3,0; 5,0; 10,0; 20,0; 30,0 y 40,0 unidades. Para aumentar el brillo de las soluciones azules y mejorar la estabilidad de los patrones, añadir 1,0 ml de solución de CuSO4 (6.3.6) a cada tubo. Luego añadir 5,0 ml de tampón de solución de color (6.3.3) y diluir con agua hasta 10,0 ml, añadir 4 gotas (0,08 ml) de la solución BQC (6.3.5) con agua hasta 10,0 ml, añadir 4 gotas (0,08 ml) de la solución BQC (6.3.5), mezclar y dejar desarrollar el color azul 30 minutos a temperatura ambiente.
Leer las intensidades de color en el fotómetro con filtro de 610 nm, restar el valor de la prueba en blanco del color de cada patrón de fenol y preparar la curva patrón (debe ser una línea recta).
Si los patrones han de usarse para comparación visual, guardar en refrigerador. Preparar semanalmente una serie nueva.
6.4 Procedimiento.
Tomar la muestra por debajo de la superficie con cuchillo y espátula limpios y proceder como sigue:
Pesar 1,0 g de muestra (preferiblemente por duplicado) sobre un pedazo de papel encerado de aproximadamente 1 pulgada cuadrada e introducir el papel con la muestra dentro del tubo. Análogamente, pesar otra muestra y colocarla en un tubo como control o patrón.
Calentar el patrón aproximadamente 1 minuto a 85-90° en vaso de agua hirviendo (cubierto así el tubo entero se calienta a 85-90°) y se enfría a temperatura ambiente. A partir de este momento, tratar en la misma forma el patrón y el problema.
Añadir 10,0 ml de sustrato patrón (6.3.4). Tapar el tubo y mezclar. Inmediatamente después de añadir el sustrato, incubar 1 hora en baño de agua a 37-38°, mezclando o agitando el contenido de cuando en cuando.
Calentar en vaso de agua hirviendo casi 1 minuto, calentando hasta 85-90° (utilizar termómetro en otro tubo del mismo tamaño y forma que contenga el mismo volumen de líquido) y enfriar a temperatura ambiente en recipiente de agua fría.
Pipetar en 1 ml de solución de Zn SO4H2O de 6,0 g/100 ml y mezclar por completo (el pH de la mezcla debe ser de 9,0-9,1).
Filtrar (se recomienda embudo de 5 cm y papel Whatman número 42 o núm. 2) y recoger 5,0 ml de filtrado en el tubo, preferentemente graduado a 5,0 y 10,0 ml.
Añadir 5,0 ml de tampón de desarrollo de color (6.3.2). El pH de la mezcla ha de ser 9,3-9,4.
Añadir 4 gotas de la solución BQC (6.3.5) y dejar 30 minutos a temperatura ambiente para desarrollo de color (para detectar únicamente la pasteurización añadir solamente 2 gotas de solución BQC).
Determinar la intensidad del color azul por uno de los siguientes métodos:
a) Con fotómetro.‒Leer intensidades de color de soluciones en blanco y problema (utilizando filtro con transmitancia máxima a aproximadamente 610 nm, restar la lectura de la prueba en blanco de la del problema, y expresar el resultado en equivalentes fenol mediante referencias a la curva patrón obtenida con las correspondientes soluciones (6.3.8.2). Generalmente es innecesaria la extracción con alcohol butílico cuando se utiliza el fotómetro; si se hace la extracción con alcohol butílico como en (b), centrifugar la muestra 5 minutos para romper la emulsión y separar el agua suspendida en la capa de alcohol (para esta finalidad puede adaptarse una centrífuga Babcock haciendo adaptadores especiales para tubos en la forma siguiente: Cortar una sección de 1/4" de grueso de un tapón de goma de diámetro adecuado, que ajuste en el fondo del vaso de centrifugación. Pegar dos tapones de corcho de diámetro adecuado, perforar en el centro un orificio de dimensiones adecuadas para alojar un tubo ajustadamente e introducir la sección doble de corcho en el vaso. Después de centrifugar, quitar casi todo el alcohol butílico con pipeta provista de pera de goma en el extremo superior. Filtrar dentro de la cubeta del fotómetro y leer con filtro cuyo máximo de transmitancias es aproximadamente de 650 m/m).
b) Con patrones visuales.‒Con muestras que producen más de 5 unidades, comparar colores en tubos con los de patrones de fenol en solución acuosa (6.3 8.2). Para cuantificar resultados en los casos dudosos (p ej., problemas que producen 0,5-5 unidades de color) extraer con alcohol butílico (6.3.7). Añadir 5,0 ml de alcohol (6.3.7) e invertir el tubo lentamente varias veces; centrifugar como en (a) si es necesario incrementar la transparencia de la capa de alcohol, y comparar el color azul con los colores de los patrones de fenol (6.3.8.2), análogamente tratados.
En los problemas que se consideren muy positivos durante el desarrollo de color (p. ej., 20 unidades), en los que 4 gotas de solución BQC (6.3.5) pueden ser insuficientes para combinar con todo el fenol, pipetar proporción adecuada de contenidos dentro de otro tubo, diluir hasta 10,0 ml con tampón de dilución de color (6.3.3), y añadir 2 gotas adicionales de solución BQC (6.3.5). Con cada problema diluir y tratar la prueba en blanco análogamente. Si la prueba sobre la muestra diluida es todavía muy fuertemente positiva, diluir de nuevo en la misma forma hasta que el color final esté dentro del intervalo de los patrones visuales o de la curva patrón del fotómetro. Dejar 30 minutos para el desarrollo de color después de la última adición de la solución de BQC (6.3.5) antes de hacer la lectura final. Para corregir lecturas por dilución, multiplicar por 2 para dilución 5 + 5, por 10 para dilución 1 9 y por 50 dilución 1 + 9 seguida de dilución 2 + 8, etc.
6.5 Cálculo.
Cuando se utilice 1,0 g de mantequilla y se añadan 11,0 ml de líquido, multiplicar el valor de la lectura por 1,1 para convertir el resultado en equivalentes de fenol/0,5 g de mantequilla (valores mayores de 2 equivalentes/0,5 g de mantequilla indican pasteurización insuficiente).
6.6 Referencia.
1. A. O. A. C. Official Methods of Analysis, 11.ª Ed. (1970).
7. ÍNDICES DE ÁCIDOS GRASOS VOLÁTILES SOLUBLES E INSOLUBLES
7.1 Principio.
El índice de ácidos grasos volátiles solubles (índice de Reichert o Reichert-Meissl-Mellny) es el número de ml de una solución acuosa de álcali 0,1 N, requerido para neutralizar los ácidos grasos volátiles solubles en agua de 5 g de grasa en las condiciones que se especifican.
El índice de ácidos grasos volátiles insolubles (índice de Polenske) es el número de ml de solución acuosa de álcali 0,1 N, requerido para neutralizar los ácidos grasos volátiles insolubles en agua, obtenidos en 5 g de grasa, en las condiciones especificadas en el método.
Después de saponificar la grasa con una solución de hidróxido sódico en glicerina, la solución jabonosa se diluye con agua y se acidifica con ácido sulfúrico. Los ácidos grasos volátiles se destilan y los ácidos grasos insolubles se separan de los solubles por filtración. La solución acuosa de ácidos solubles y la solución etanólica de ácidos insolubles se valoran separadamente con una solución de álcali normalizada.
El método es empírico porque sólo determina una parte de estos ácidos. Por tanto, las especificaciones referentes al procedimiento y aparatos se deben seguir rigurosamente para obtener resultados exactos y reproducibles.
7.2 Material y aparatos (figura 7.1).
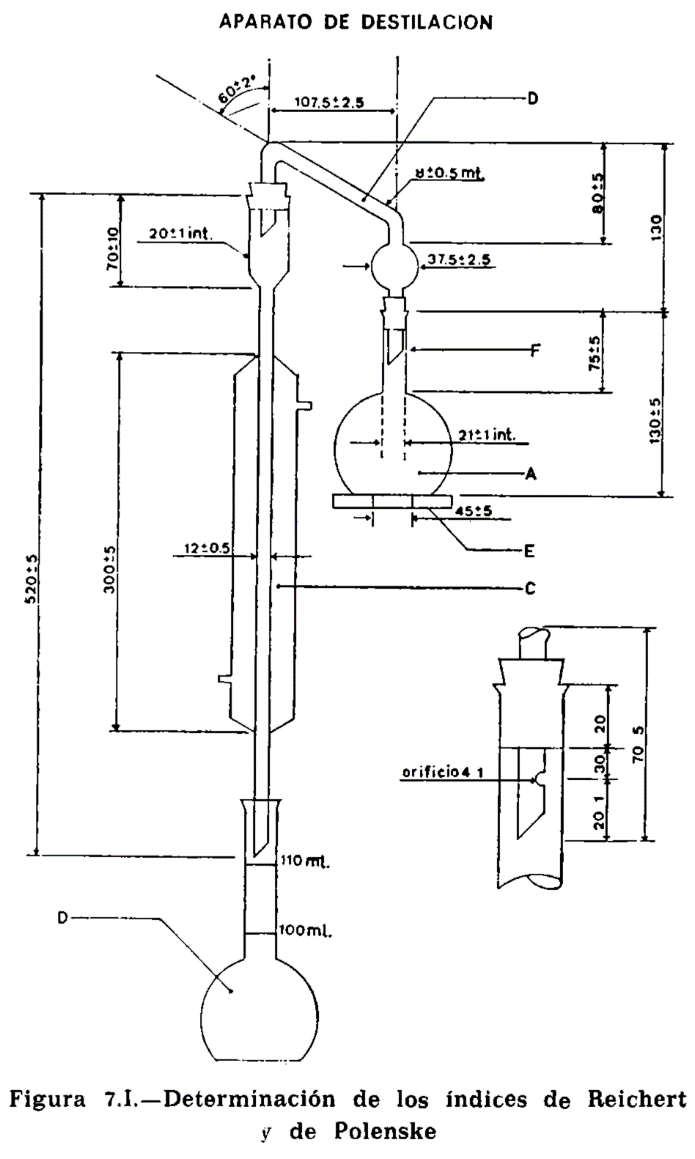
7.2.1 Matraz de fondo plano de vidrio al borosilicato de 300 ml de capacidad (A).
7,2.2 Cabeza de destilación (E).
7.2.3 Refrigerante (C).
7.2.4 Receptor, que consiste en un matraz aforado con las rayas circulares de aforo a 100 y 110 ml (D).
7.2.5 Lámina de asbesto de 120 mm de diámetro, 6 mm de espesor con una abertura central circular de 40 a 50 mm de diámetro, para sostener el matraz durante el calentamiento (E).
7.2.6 Piedra pómez triturada que pasa a través de un tamiz de malla circular de 1,44 min.
En la figura se representan las dimensiones en mm y el montaje del aparato de destilación; para las conexiones se puede utilizar tapones de caucho, neoprano o silicona, o juntas de vidrio esmerilado «estandar» 24/40.
7.3 Reactivos.
7.3.1 Glicerina (d = 1,26; 98 por 100 p/p).
7.3.2 Solución acuosa de hidróxido sódico (44 por 100 p/p), conservada en botella protegida del dióxido de carbono. Usar la porción limpia libre de precipitado de carbonatos.
7.3.3 Agua destilada, hervida durante 15 minutos, para eliminar el dióxido de carbono.
7.3.4 Solución de ácido sulfúrico 1 N.
7.3.5 Solución acuosa de hidróxido sódico 3 potásico 0,1 N, exactamente normalizada.
7.3.6 Solución indicadora de fenolftaleína (1 por 100 en etanol de 95-96 por 100).
7.3.7 Etanol (95-96 por 100 v/v) neutro a la fenolfteína. El agua usada debe ser destilada o de una pureza por lo menos equivalente.
7.4 Procedimiento.
7.4.1 Preparación de la muestra. Como en 5.4.1.
7.4.2 Determinación del índice de ácidos grasos volátiles solubles: Pesar 5 g con aproximación de 0,01 g de grasa en el matraz A. Añadir 20 g (16 ml de glicerina y 2 ml de solución de hidróxido sódico (44 por 100). Para añadir la solución de hidróxido sódico, usar una bureta protegida de la entrada de dióxido de carbono y limpiar la punta de la bureta desechando las primeras gotas. Calentar el matraz a fuego directo, evitando sobrecalentar y agitando continuamente, hasta que el líquido no forme espuma y se vuelva límpido Dejar enfriar el matraz hasta 90 °C. Añadir 90 ml de agua destilada recientemente hervida a la misma temperatura aproximadamente y mezclar. El líquido debe quedar límpido. Añadir de 0,6 a 0,7 g de piedra pómez y después 50 ml de solución de ácido sulfúrico 1 N. Conectar inmediatamente el matraz al aparato de destilación y calentarlo ligeramente hasta que los ácidos grasos libres formen una capa superficial limpia. Empezar a calentar y regular la llama de modo que se recojan en el matraz aforado 110 ml de destilado en 19-21 minutos, tomando como principio del período de destilación el momento en que se forma la primera gota en el refrigerante. Regular el flujo de agua del refrigerante de modo que se mantenga la temperatura del agua que sale del refrigerante a 20 ± 1 °C.
Cuando se hayan recogido exactamente 110 ml de destilado, quitar el mechero inmediatamente y sustituir el matraz aforado por un pequeño vaso. Mezclar el contenido del matraz aforado agitando suavemente y sumergir el matraz en un baño de agua a 20 ± 1 °C durante 10-15 minutos, estando la señal de 110 ml del matraz aforado por debajo del nivel del agua del baño. Agitar el matraz de cuando en cuando. Tapar el matraz y mezclar invirtiéndolo 4 ó 5 veces sin agitar. Filtrar los 110 ml de destilado por un papel filtro seco de velocidad media (diámetro 80-90 mm), que se ajusta cómodamente en un embudo. El filtrado debe ser límpido (el filtro debe ser de tales dimensiones, que un volumen de 15 ml lo llene completamente). Pipetar 100 ml de filtrado y pasarlos a un matraz erlenmeyer de 300 ml. añadir 0,5 ml de la solución indicadora de fenolftaleína y valorar con la solución acuosa de álcali «estandar» 0,1 N hasta un color rosa persistente durante 0,5-1 minuto.
7.4.3 Ensayo en blanco: Hacer un ensayo en blanco sin grasa y en lugar de saponificar a fuego directo calentar en baño de agua hirviendo durante 15 minutos.
No se requerirán para la, valoración más de 0,5 ml de la solución de álcali normalizada. En otro caso, se deben preparar nuevas soluciones del reactivo.
7.4.4 Determinación del índice de ácidos grasos volátiles insolubles (Polenske): Lavar el filtro con tres porciones sucesivas de 15 ml de agua destilada a la temperatura de 20 ± 1 °C, habiendo pasado previamente cada una a través del refrigerante del vaso pequeño y del matraz aforado. Poner el embudo y el filtro en el cuello de un matraz cónico, limpio y seco, de 200 ml de capacidad. Disolver los ácidos grasos insolubles repitiendo los lavados, usando ahora porciones de 15 ml de etanol (95-96 por 100 v/v). Valorar con la solución acuosa de álcali normalizada (0,1 N) el conjunto de los lavados con etanol usando 0,5 ml de solución indicadora de fenolftaleína, hasta un color rosa persistente durante 0,5-1 minuto.
7.5 Cálculo.
7.5.1 Índice de ácidos grasos volátiles solubles (índice de Reichert).
Índice de Reichert = 11 · t · (V1 ‒ b)
Siendo:
V1 = volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en la valoración de la muestra.
b = volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en el ensayo en blanco.
t = normalidad exacta de la solución normalizada (0,1 N) de álcali.
Redondear el resultado a la primera cifra decimal.
7.5.2 Índice de ácidos grasos volátiles insolubles (índice de Polenske).
Índice de Polenske = 10 ∙ t · V2
Siendo:
V2 = volumen en mililitros de la solución normalizada (0,1 N) de álcali utilizada en la valoración de la muestra.
t = normalidad exacta de la solución normalizada (0,1 N) de álcali.
Redondear el resultado a la primera cifra decimal.
7.5.3 Reproducción de resultados: La diferencia entre los resultados de dos determinaciones duplicadas (resultados obtenidos simultáneamente o inmediatamente uno detrás de otro por el mismo analista) no debe exceder de 0,5 para el índice de Reichert o de 0,3 para el índice de Polenske.
7.6 Referencia.
1. Norma Internacional FIL-IDF 37: 1966.
8. ÍNDICE DE KIRSCHNER
8.1 Método operatorio.
1.° Neutralizar 100 ml del destilado Reichert-Meissl con solución de Ba (OH)2 0,1 N, con toda precisión hasta lograr un débil color rosado empleando 0,5 ml del indicador. Realizar la titulación en un matraz cerrado para evitar la absorción de CO2.
2.º Añadir 0,3 g de Ag2SO4, en forma de polvo fino. Dejar reposar la mezcla una hora, agitando con frecuencia y filtrándola después.
3.º Recoger 100 ml del filtrado, colocarlo en un frasco de destilación de 300 ml, añadir 35 ml de agua destilada y 10 ml de H2SO4 diluido. Añadir un trozo de alambre de aluminio o varios trozos de piedra pómez para evitar que el líquido rebose. Únase al destilador y comiéncese la destilación a la velocidad de 110 ml en unos 20 minutos.
4.º Después de recoger 110 ml del destilado, filtrar esta cantidad total y titular 100 ml con NaOH, 1 N, empleando 0,5 ml del indicador hasta lograr un tono rosado que persista durante 2-3 minutos.
5.º Preparar y realizar una prueba en blanco semejante a la anterior en todos sus aspectos.
8.2 Cálculo.

A = titulación de la muestra ‒ titulación en blanco.
B = volumen, en ml, de Ba (OH)2 0,1 N, requeridos para neutralizar los 100 ml originales del destilado de Reichert-Meissl.
8.3 Referencia.
1. Norma Internacional AOCS 5-40.
Vinos
1. EXAMEN ORGANOLÉPTICO
1.1 Examen del color.
Determinar mediante apreciación visual las características, bien definidas, del color del vino.
Observar el vino colocado en copas de cristal fino e incoloro, o bien en tazas de plata o de fondo plateado, el cual presentará diferentes formas convexas o abolladas, para apreciar el vino en diferentes espesores y sobre fondo brillante.
Utilizar las siguientes calificaciones del color para vinos tintos: rojo rubí, rojo violáceo o morado, viraje a tono cebolla o amarillento.
Según la intensidad del color, el vino tinto puede calificarse en: vino tinto, rosado, clarete, y si tiene mucho o poco color dentro de estos tipos, se dirá que tiene mucha o poca capa.
En vinos blancos, utilizar las calificaciones de tono verdoso, casi incoloro, amarillo, amarillo-oro, ambarino, pajizo claro u oscuro (rancia), etc., y en todos los tipos, si el color es blanco, o si azulea, pardea o ennegrece.
1.2 Formación de espuma.
Observar el aspecto de la espuma que se forma al agitar el vino en la copa y determinar si es o no abundante, si desaparece rápidamente o, en caso de vinos tintos, si persiste unos momentos el color más o menos intenso. El vino de mucha capa o el vino enyesado produce espuma que tarda mucho en desaparecer.
1.3 Presencia de gas carbónico.
Verter vino en una copa. La formación de burbujas indica la presencia de gas carbónico en el vino. Este gas carbónico debe eliminarse para continuar el examen de la muestra, agitando o en la forma que se indica en los métodos analíticos.
1.4 Limpieza.
Apreciar en copa o en taza de plata, al mismo tiempo que el color, si el vino está turbio, claro o brillante.
Observar también si hay depósito y si al agitar tiene aspecto cristalino o pulverulento que enturbie el vino.
1.5 Degustación o cata.
Proceder a la degustación recién destapada la botella, consignando datos, referentes a calidad, origen, edad y posibles alteraciones del vino.
Coger una copa de cristal ovoide por su platillo con los dedos pulgar, índice y central de la mano derecha, quedando la copa libre para examinar el vino. Llenar con vino sólo 1/3 ó 1/4 de la copa., para poder agitar bien y tener un espacio vacío donde se concentren los aromas. Agitar haciendo girar el vino y apreciar el aroma.
Apreciar y caracterizar el olor a «vinoso», que acusa vino nuevo, el «afrutado», el de vino que comenzó la «crianza», la riqueza alcohólica, olores ácidos volátiles, olores que acusen defectos de elaboración, etc., y la presencia de enfermedades y alteraciones.
En degustación de más precisión, como será, la necesaria para definir características de un tipo de vino y que han de servir de referencia, es necesaria reunir un grupo determinado de buenos catadores, y realizar un número de catas suficientes para obtener resultados satisfactorios estadísticamente según las normas específicas existentes.
1.6 Referencia.
1. Amerine, M. A., y Feduchy, E.: «Los resultados de la cata del vino y del análisis químico». Boletín I. N. I. A., 31, 353-375, 1956.
2. ENSAYOS PREVIOS DE CONSERVACIÓN
2.1 Prueba del aire.
2.1.1 «Quiebras» tánico-férricas o fosfato-férricas.‒Dejar el vino en contacto con el aire (en una copa mediada), en sitio fresco y al resguardo de la luz. Observar al cabo de 12 o más horas. El enturbiamiento o el ennegrecimiento pone de manifiesto la posibilidad de «quiebras» tánico-férricas o fosfato-férricas.
2.1.2 «Quiebra oxidásica».‒Proceder como en 2.1.1, utilizando dos muestras de vino, una pasterizada, para destruir la oxidasa, y la otra sin pasterizar. La formación de color pardo y precipitado pulverulento oscuro son señales características de la «quiebra oxidásica».
2.1.3 Quiebra cuprosa».‒Poner la muestra en botella cerrada, calentar en baño de agua a 35º, y exponer a la luz. Si la riqueza en cobre es suficiente, aparece enturbiamiento característico de la «quiebra cuprosa».
2.2 Prueba del frío.
Enfriar el vino a la temperatura que normalmente se suponga va a estar expuesto, y comprobar si se produce precipitación de bitartrato.
2.3 Prueba de la estufa.
Mantener una muestra de vino en estufa a 22-25° durante 3-4 días. Examinar al microscopio el posible desarrollo microbiano distinguiendo si se trata de levaduras o de bacterias. Si es necesario, proceder al conteo y la identificación de los microorganismos.
2.4 Examen microscópico.
Realizar el examen microscópico sobre preparación del poso de la muestra, que puede aparecer después de reposo en estufa o a temperatura ambiente.
Si el vino se presenta turbio en el envase original, hacer una preparación directamente del vino o del poso obtenido por centrifugación de ese vino. Si no se presenta depósito y el vino está poco turbio, hacer una preparación del poso obtenido por centrifugación.
Determinar en el examen microscópico si el depósito es microbiano, distinguiendo levaduras y bacterias.
3(a). COLOR DE LOS VINOS
3(a).1 Principio.
El color de los vinos se determina por transparencia como se percibe por la vista, pero por un procedimiento independiente de la apreciación personal, valiéndose de métodos espectrofoto métricos triestimulares de ordenadas seleccionadas de Hardy, fundado en el sistema de la Commision Internationale de l'Eclairage (C. I. E.), con relación a la luz producida por un cielo nublado (fuente C).
3(a).2 Material yaparatos.
3(a).2.1 Espectrofotómetro para medida en el espectro visible. Los valores de transmitancia correspondientes a una misma muestra no deben acusar diferencias superiores a 0,005, y cuando la escala del aparato esté graduada en valores de transmitancia multiplicados por 100, no debe haber diferencia superior a 0,5.
Las cubetas serán de cuarzo, de paredes paralelas y espesor interno b, que se expresa en centímetros, y con una aproximación de ± 0,002.b.
Conviene disponer de cuatro pares de cubetas en las que los espesores b sean de 0,1 cm, 0,2 cm, 0,5 cm y 1 cm.
Según la intensidad del color se escogerán un par de cubetas de tal forma que la absorbancia A quede comprendida entre 0,3-0,7 (transmitancia 0,5-0,2).
3(a).3 Procedimiento.
Si el vino no está limpio, centrifugar previamente. Eliminar el gas carbónico, si es necesario, por agitación con vacío parcial.
Medir directamente con el espectrofotómetro las transmitancias del vino a las cuatro longitudes de onda = 625, 550, 495 y 445 mμ, empleando la cubeta de espesor conveniente, según intensidad del color del vino.
3(a).4 Cálculo y expresión de los resultados.
Utilizar agua destilada como líquido de referencia.
3(a).4.1 Calcular las coordenadas (x, y) del punto representativo del color del vino en el diagrama tricromático de la C. I. E.


X = 0,42 ∙ T625 ‒ 4 ‒ 0,35 ∙ T550 + 0,21 ∙ T445.
Y = 0,20 ∙ T625 + 0,63 ∙ T550 + 0,17 ∙ T495.
Z = 0,24 ∙ T495 + 0,94 ∙ T445.
Los valores triestimulares X, Y, Z expresan las proporciones de colores rojos, verde y azul que dan por mezcla el color del vino.
Cuando el espesor o de la cubeta sea inferior a 1 cm, referir la transmitancia a 1 cm en la siguiente forma: T = T1/b, y si la transmitancia viene expresada en porcentaje

T = transmitancia referida a 1 cm de espesor de cubeta.
T' = transmitancia obtenida para b cm de espesor de cubeta.
b = espesor en cm de la cubeta utilizada.
3(a).4.2 Luminosidad relativa.‒Es el valor de Y, expresado en porcentaje (siendo el negro Y = 0 y el incoloro Y = 100).
3(a).4.3 Cromaticidad.‒El color del vino por transparencia a la fuente C con el espesor de 1 cm viene dado por el punto del diagrama de cromaticidad de la C. I. E. (fig. 3(a),I), definido por las coordenadas x e y.
3(b). COLOR DE LOS VINOS
(Aplicable a vinos tintos y rosados)
3 (b).1 Principio.
La intensidad de color se mide por la suma de las absorbencias del vino para un espesor de 1 cm, correspondientes a las longitudes de onda de absorbancia mínima (420 mμ) del vino tinto.
La tonalidad se expresa por el ángulo que forma con el eje de longitudes de onda la cuerda que une los puntos de la curva espectrofotométrica representativos de las absorbancias correspondientes a las longitudes de onda de 420 y 520 mμ.
3(b).2 Material y aparatos.
Como en 3(a).2.
3(b).3 Procedimiento.
Como en 3(a).3, excepto que las longitudes de onda utilizadas serán de 420 mμ y 520 mμ.
3(b).4 Cálculos.
Calcular la intensidad colorante y la tonalidad.
3(b).4.1 Intensidad colorante:

A420 = absorbancia a 420 mμ.
A520 = absorbancia a 520 mμ.
b = espesor en cm de la cubeta.
3(b).4.2 Tonalidad.‒La tonalidad se mide por el ángulo cuya tangente es igual a la diferencia del valor numérico de las dos absorbancias A520 ‒ A420. Conocido este valor, se puede determinar el valor del ángulo por tablas expresadas en grados sexagesimales.
3(b).5 Observaciones.
Con este método no se determina el color, sino la intensidad y la tonalidad del color, que son características cromáticas convencionales. Por su rapidez y sencillez, es muy práctico para comprobar la evolución de la materia colorante durante su «crianza» o añejamiento.
3(b).6 Referencias.
1. Determinación triestimular del color de los vinos, método simplificado por C. Stella. Comunicación al O. I. V. núm. 282 (2 feb. 1968).
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. AO, 1-14. 1969.
4(a). MASA VOLÚMICA Y DENSIDAD RELATIVA
(Método picnométrico)
4(a).1 Principio.
La masa volúmica es el cociente de la masa de un cierto volumen de vino o de mosto por este volumen. Se expresa en g por ml y su símbolo es ρ20.
La densidad relativa es el cociente de la masa volúmica del vino por la masa volúmica del agua. Su símbolo es d20 ó simplemente d, cuando no haya posibilidad de confusión.
La masa volúmica y la densidad relativa de un vino se determina a 20°.
El método picnométrico consiste en la determinación ponderal del contenido en alcohol de un destilado por medio de un picnómetro.
4(a).2 Material y aparatos.
4(a).2.1 Picnómetro de vidrio de 50 ml de capacidad, de cuello con diámetro interior de 3,5 mm. Embudo y sifón para picnómetros.
4(a).2.2 Termómetro contrastado dividido en 1/5 ‒ 1/10 de grado Celsius graduado de 10 a 30 °C.
4(a).2.3 Termostato regulado a 20 ± 0,2 °C.
4(a).2.4. Balanza con aproximación de 0,1 mg.
4(a).3 Procedimiento.
Lavar bien el picnómetro y enjuagarlo después con alcohol y luego con éter, escurrir bien y secar cuidadosamente con paño de hilo o papel filtro el exterior, y con corriente de aire seco el interior, tapar, dejar en reposo en la caja de la balanza durante 30 minutos aproximadamente, y pesar.
Llenar después con agua a 20° recién destilada, con cuidado de evitar burbujas de aire en el interior del picnómetro. Sumergir en el agua a la temperatura comprobada de 20°.
Mantener el picnómetro en el termostato durante 30 minutos, y enrasar el nivel del agua con la marca del cuello. Tapar el picnómetro, secar exteriormente con las precauciones expuestas, dejar nuevamente 30 minutos en la caja de la balanza y después pesar.
Vaciar el picnómetro, enjuagar nuevamente, primero con agua y después con alcohol y éter, y secar como anteriormente, llenar con el vino a temperatura de 20 °C, aproximadamente, cuidando que no quede ninguna burbuja de aire (en caso de que el vino tenga carbónico, se eliminará previamente como luego se indica). Dejar el picnómetro con el vino en el termostato a 20 °C ± 0,2° durante 30 minutos, llenar hasta volumen con el vino y pesar.
4(a).4. Cálculo.
4(a).4.1 Cálculo de la densidad relativa a 20 °C.

P = peso en g del picnómetro vacío.
P' = peso en g del picnómetro más agua a 20 °C.
P" = peso en g del picnómetro más vino a 20 °C.
4(a).4.2 Cálculo de la masa volúmica ρ20.

o bien

El valor de c se encuentra en las tablas 4(a),I y 4(a),II.
La aproximación de los resultados será de 0,0003. Para mayor precisión, se tendrá en cuenta el empuje del aire como en 5(a).5.
4(a).5 Observaciones.
Si el vino contiene cantidad sensible de gas carbónico, se eliminará todo lo posible agitando un volumen de unos 250 mililitros del vino, en matraz de 1.000 ml.
Si el vino está turbio, se filtrará por papel de filtración rápida, de pliegues y procurando airear lo menos posible, tapando también el embudo con vidrio de reloj.
Si el enturbiamiento es por suspensión de levadura, la filtración corriente aludida no lo aclarará, siendo conveniente en este caso, dejar decantar el vino en probeta tapada, durante dos o tres días en sitio fresco.
4(a).6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins O. I. V., Al, 11-26. 1969.
TABLA 4(a).I
Cálculo de la masa volúmica (ρ20) a partir de la densidad relativa 20/20 y recíprocamente
| Densidades | 0,960 | 0,980 | 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 |
| Correcciones c | 1,73 | 1,76 | 1,80 | 1,84 | 1,87 | 1,91 | 1,94 | 1,98 |
| Densidades | 1,120 | 1,140 | 1,160 | 1,180 | 1,200 | 1,220 | 1,240 | 1,260 |
| Correcciones c | 2,02 | 2,05 | 2,09 | 2,12 | 2,16 | 2,20 | 2,23 | 2,27 |
| Densidades | 1,280 | 1,300 | 1,320 | 1,340 | 1,360 | 1,380 | 1,400 | |
| Correcciones c | 2,30 | 2,34 | 2,38 | 2,41 | 2,45 | 2,48 | 2,52 |
TABLA 4(a).II
Cálculo de la masa volúmica (ρ16) a partir de la densidad relativa 15/15 y recíprocamente
| Densidades | 0,960 | 0,980 | 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 |
| Correcciones c | 0,86 | 0,88 | 0,90 | 0,92 | 0,94 | 0,95 | 0,97 | 0,99 |
| Densidades | 1,120 | 1,140 | 1,160 | 1,180 | 1,200 | 1,220 | 1,240 | 1,280 |
| Correcciones c | 1,01 | 1,03 | 1,04 | 1,06 | 1,08 | 1,10 | 1,12 | 1,13 |
| Densidades | 1,280 | 1,300 | 1,320 | 1,340 | 1,360 | 1,380 | 1,400 | |
| Correcciones c | 1,15 | 1,17 | 1,19 | 1,21 | 1,22 | 1,24 | 1,25 |
4(b). MASA VOLÚMICA Y DENSIDAD RELATIVA
(Método areométrico)
4(b).1 Principio.
Determinación de la masa volúmica a partir de la lectura del areómetro que flota introducido en el vino.
4(b).2 Material y aparatos.
4(b).2.1 Areómetro. Los areómetros empleados deben responder a las siguientes normas (I. S. O.):
Parte cilíndrica sumergible y tallo de sección circular de 3 mm de diámetro como mínimo. Para vinos secos, la graduación será de 0,983 a 1,003 en milésimas y 0,2 milésimas. Las divisiones de milésimas deben ser de 5 mm como mínimo. Para vinos desalcoholizados y vinos dulces, y eventualmente para mostos, se recomienda un juego de 5 areómetros graduados de 1,000-1,030; 1,030-1,060; 1,060-1,090; 1,090-1,120; 1,120-1,150. Estos aparatos estarán graduados en masas volúmicas a 20°, en milésimas y 0,5 milésimas por lo menos. Cada división de una milésima será de 3 mm como mínimo. En los aparatos se indicará que las lecturas deben hacerse por la parte superior del menisco.
4(b).2.2 Termómetro contrastado graduado por lo menos en 1/2 grados.
4(b).2.3 Probeta cilíndrica de 36 mm de diámetro interior y 320 mm de altura.
4(b).2.4 Plataforma con tornillos de nivelación, para mantener vertical la probeta sobre ella colocada.
4(b).3 Procedimiento.
Colocar en la probeta descrita 250 ml del vino e introducir el areómetro y el termómetro.
El areómetro y especialmente el tallo graduado deberán estar limpios y desengrasados.
Agitar para uniformar la temperatura. Un minuto después, hacer la lectura del termómetro. Retirar el termómetro y hacer la lectura de la masa aparente sobre el tallo del areómetro.
4(b).4 Cálculo.
Calcular la masa volúmica referida a 20 °C.

(‒ si t es inferior a 20 °C)
(+ si t es superior a 20 °C)
ρ20 = masa volúmica referida a 20°.
ρt = lectura del tallo graduado del areómetro.
c = valor de la corrección correspondiente a la temperatura del vino en el momento de la lectura. Este valor se encuentra en las tablas 4(b).1 y 4(b),II.
t = temperatura del vino en el momento de la lectura. Los resultados se obtendrán con una precisión de 0,0003.
4(b).5 Observaciones.
Si se utilizan areómetros que den  ó
ó  , hay que corregir la lectura en las tablas correspondientes, y después expresar los resultados en
, hay que corregir la lectura en las tablas correspondientes, y después expresar los resultados en  ó ρ20, siguientes:
ó ρ20, siguientes:




4(b).6 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. o. I. V. A1, 11-26.
2. P. Jaulmes, S. Brun: «Tables de correspondance entre les diverses tables Alcoométriaues». Société Pharmacia Montpellier, 26: 111,141, 1966.
TABLA 4(b).I
Correcciones para referir la masa volúmica de los vinos secos a 20°
| Temperaturas | GRADOS ALCOHÓLICOS | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | |
| 10º | 1,45 | 1,51 | 1,55 | 1,58 | 1,64 | 1,70 | 1,78 | 1,88 | 1,98 | 2,09 | 2,21 | 2,34 | 2,47 | 2,60 | 2,75 | 2,90 | 3,06 | 3,22 | 3,39 | 3,57 | 3,75 | 3,93 | 4,12 | 4,31 |
| 11º | 1,35 | 1,40 | 1,43 | 1,47 | 1,52 | 1,58 | 1,65 | 1,73 | 1,83 | 1,93 | 2,03 | 2,15 | 2,26 | 2,36 | 2,51 | 2,65 | 2,73 | 2,93 | 3,08 | 3,24 | 3,40 | 3,57 | 3,73 | 3,90 |
| 12° | 1,24 | 1,28 | 1,31 | 1,34 | 1,39 | 1,44 | 1,50 | 1,58 | 1,66 | 1,75 | 1,84 | 1,94 | 2,04 | 2,15 | 2,20 | 2,38 | 2,51 | 2,63 | 2,77 | 2,91 | 3,05 | 3,19 | 3,34 | 3,49 |
| 13° | 1,12 | 1,16 | 1,18 | 1,21 | 1,25 | 1,30 | 1,35 | 1,42 | 1,49 | 1,56 | 1,64 | 1,73 | 1,82 | 1,91 | 2,01 | 2,11 | 2,22 | 2,33 | 2,45 | 2,57 | 2,69 | 2,81 | 2,05 | 3,07 |
| 14° | 0,99 | 1,03 | 1,05 | 1,07 | 1,11 | 1,14 | 1,19 | 1,24 | 1,31 | 1,37 | 1,44 | 1,52 | 1,59 | 1,67 | 1,75 | 1,84 | 1,93 | 2,03 | 2,13 | 2,23 | 2,33 | 2,44 | 2,55 | 2,66 |
| 15° | 0,86 | 0,89 | 0,90 | 0,92 | 0,95 | 0,93 | 1,02 | 1,07 | 1,12 | 1,17 | 1,23 | 1,29 | 1,35 | 1,42 | 1,49 | 1,56 | 1,63 | 1,71 | 1,80 | 1,88 | 1,96 | 2,05 | 2,14 | 2,23 |
| 16° | 0,71 | 0,73 | 0,74 | 0,76 | 0,78 | 0,31 | 0,84 | 0,87 | 0,91 | 0,93 | 0,99 | 1,03 | 1,10 | 1,15 | 1,21 | 1,27 | 1,33 | 1,39 | 1,45 | 1,52 | 1,59 | 1,66 | 1,73 | 1,80 |
| 17° | 0,55 | 0,37 | 0,57 | 0,50 | 0,60 | 0,62 | 0,65 | 0,67 | 0,70 | 0,74 | 0,77 | 0,81 | 0,84 | 0,88 | 0,92 | 0,99 | 1,01 | 1,05 | 1,11 | 1,15 | 1,20 | 1,26 | 1,31 | 1,36 |
| 18° | 0,33 | 0,39 | 0,39 | 0,40 | 0,41 | 0,43 | 0,44 | 0,46 | 0,48 | 0,50 | 0,52 | 0,55 | 0,57 | 0,60 | 0,62 | 0,65 | 0,63 | 0,71 | 0,74 | 0,78 | 0,81 | 0,85 | 0,88 | 0,91 |
| 19° | 0,19 | 0,20 | 0,20 | 0,21 | 0,21 | 0,22 | 0,23 | 0,24 | 0,25 | 0,26 | 0,27 | 0,23 | 0,29 | 0,30 | 0,32 | 0,33 | 0,35 | 0,36 | 0,38 | 0,39 | 0,41 | 0,43 | 0,44 | 0,46 |
| 20° | ||||||||||||||||||||||||
| 21° | 0,21 | 0,22 | 0,22 | 0,23 | 0,23 | 0,24 | 0,25 | 0,25 | 0,26 | 0,27 | 0,29 | 0,29 | 0,31 | 0,32 | 0,34 | 0,35 | 0,36 | 0,38 | 0,39 | 0,41 | 0,43 | 0,44 | 0,46 | 0,48 |
| 22° | 0,43 | 0,45 | 0,45 | 0,46 | 0,47 | 0,49 | 0,50 | 0,52 | 0,54 | 0,56 | 0,58 | 0,60 | 0,63 | 0,65 | 0,68 | 0,71 | 0,73 | 0,77 | 0,80 | 0,83 | 0,86 | 0,89 | 0,93 | 0,96 |
| 23° | 0,67 | 0,69 | 0,70 | 0,71 | 0,72 | 0,74 | 0,77 | 0,79 | 0,82 | 0,85 | 0,88 | 0,91 | 0,95 | 0,99 | 1,03 | 1,07 | 1,12 | 1,16 | 1,21 | 1,25 | 1,30 | 1,35 | 1,40 | 1,45 |
| 24° | 0,91 | 0,93 | 0,95 | 0,97 | 0,99 | 1,01 | 1,04 | 1,07 | 1,11 | 1,15 | 1,20 | 1,24 | 1,29 | 1,34 | 1,39 | 1,45 | 1,50 | 1,56 | 1,62 | 1,69 | 1,76 | 1,82 | 1,88 | 1,95 |
| 25° | 1,16 | 1,19 | 1,21 | 1,23 | 1,26 | 1,29 | 1,33 | 1,37 | 1,42 | 1,47 | 1,52 | 1,57 | 1,63 | 1,70 | 1,76 | 1,83 | 1,90 | 1,97 | 2,05 | 2,13 | 2,21 | 2,29 | 2,37 | 2,45 |
| 26° | 1,42 | 1,46 | 1,49 | 1,51 | 1,54 | 1,58 | 1,62 | 1,57 | 1,73 | 1,79 | 1,85 | 1,92 | 1,90 | 2,07 | 2,14 | 2,22 | 2,31 | 2,40 | 2,49 | 2,56 | 2,67 | 2,77 | 2,86 | 2,96 |
| 27° | 1,69 | 1,74 | 1,77 | 1,80 | 1,63 | 1,88 | 1,93 | 1,92 | 2,05 | 2,12 | 2,20 | 2,27 | 2,35 | 2,44 | 2,53 | 2,63 | 2,72 | 2,82 | 2,93 | 3,04 | 3,14 | 3,25 | 3,37 | 3,48 |
| 28º | 1,97 | 2,03 | 2,06 | 2,09 | 2,14 | 2,19 | 2,24 | 2,31 | 2,38 | 2,46 | 2,55 | 2,63 | 2,73 | 2,83 | 2,93 | 3,03 | 3,14 | 3,26 | 3,38 | 3,50 | 3,62 | 3,75 | 3,85 | 4,00 |
| 29° | 2,26 | 2,33 | 2,37 | 2,40 | 2,45 | 2,50 | 2,57 | 2,64 | 2,73 | 2,82 | 2,91 | 2,99 | 3,11 | 3,22 | 3,34 | 3,45 | 3,59 | 3,70 | 3,84 | 3,97 | 4,11 | 4,25 | 4,39 | 4,54 |
| 30° | 2,56 | 2,64 | 2,67 | 2,72 | 2,77 | 2,83 | 2,90 | 2,98 | 3,03 | 3,16 | 3,28 | 3,33 | 3,50 | 3,62 | 3,75 | 3,88 | 4,02 | 4,16 | 4,30 | 4,46 | 4,61 | 4,76 | 4,92 | 5,07 |
TABLA 4(b).II
Correcciones para referir la masa volúmica de los vinos dulces a 20°
| Temperaturas | MASAS VOLÚMICAS | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VINOS DE 13º | VINOS DE 15º | VINOS DE 17º | |||||||||||||||||||
| 1,000 | 1,020 | 1,040 | 1,000 | 1,030 | 1,100 | 1,120 | 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,120 | 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,120 | |
| 10º | 2,24 | 2,53 | 2,93 | 3,27 | 3,59 | 3,39 | 4,13 | 2,51 | 2,35 | 3,20 | 3,54 | 3,86 | 4,02 | 4,40 | 2,81 | 3,15 | 3,50 | 3,34 | 4,15 | 4,45 | 4,74 |
| 11º | 2,06 | 2,37 | 2,09 | 2,97 | 3,26 | 3,53 | 3,78 | 2,31 | 2,61 | 2,93 | 2,21 | 3.51 | 3,64 | 4,02 | 2,57 | 2,89 | 3,20 | 3,49 | 3,77 | 4,03 | 4,28 |
| 12º | 1,87 | 2,14 | 2,42 | 2,67 | 2,94 | 3,17 | 3,40 | 2,09 | 2,36 | 2,64 | 2,90 | 3,16 | 3,27 | 3,61 | 2,32 | 2,60 | 2,87 | 3,13 | 3,39 | 3,63 | 3,84 |
| 13º | 1,69 | 1,93 | 2,14 | 2,37 | 2,59 | 2,80 | 3,00 | 1,88 | 2,12 | 2,34 | 2,56 | 2,78 | 2,83 | 3,19 | 2,09 | 2,33 | 2,55 | 2,77 | 2,98 | 3,19 | 3,39 |
| 14º | 1,49 | 1,70 | 1,90 | 2,09 | 2,27 | 2,44 | 2,61 | 1,67 | 1,06 | 2,06 | 2,25 | 2,45 | 2,51 | 2,77 | 1,83 | 2,03 | 2,23 | 2,42 | 2,61 | 2,77 | 2,94 |
| 15° | 1,25 | 1,42 | 1,59 | 1,75 | 1,90 | 2,05 | 2,19 | 1,39 | 1,56 | 1,72 | 1,83 | 2,03 | 2,11 | 2,32 | 1,54 | 1,71 | 1,37 | 2,03 | 2,13 | 2,32 | 2,47 |
| 16º | 1,03 | 1,17 | 1,30 | 1,43 | 1,55 | 1,67 | 1,73 | 1,06 | 1,27 | 1,40 | 1,53 | 1,65 | 1,77 | 1,88 | 1,25 | 1,39 | 1,52 | 1,65 | 1,77 | 1,89 | 2,00 |
| 17° | 0,80 | 0,90 | 1,00 | 1,00 | 1,17 | 1,27 | 1,30 | 0,87 | 0,93 | 1,00 | 1,17 | 1,26 | 1,35 | 1,44 | 0,96 | 1,06 | 1,16 | 1,26 | 1,35 | 1,44 | 1,52 |
| 18º | 0,54 | 0,61 | 0,68 | 0,75 | 0,31 | 0,86 | 0,92 | 0,60 | 0,66 | 0,73 | 0,80 | 0,66 | 0,91 | 0,97 | 0,66 | 0,72 | 0,79 | 0,86 | 0,92 | 0,97 | 1,03 |
| 19º | 0,29 | 0,33 | 0,36 | 0,39 | 0,42 | 0,45 | 0,48 | 0,32 | 0,36 | 0,39 | 0,42 | 0,45 | 0,43 | 0,51 | 0,35 | 0,38 | 0,41 | 0,45 | 0,43 | 0,51 | 0,53 |
| 20º | |||||||||||||||||||||
| 21º | 0,29 | 0,32 | 0,35 | 0,38 | 0,42 | 0,45 | 0,47 | 0,32 | 0,35 | 0,33 | 0,42 | 0,45 | 0,43 | 0,50 | 0,34 | 0,33 | 0,41 | 0,44 | 0,47 | 0,59 | 0,53 |
| 22º | 0,57 | 0,64 | 0,70 | 0,76 | 0,32 | 0,38 | 0,93 | 0,98 | 0,69 | 0,75 | 0,81 | 0,87 | 0,93 | 0,93 | 0,68 | 0,75 | 0,81 | 0,27 | 0,93 | 0,99 | 1,04 |
| 23º | 0,89 | 0,98 | 1,08 | 1,17 | 1,26 | 1,34 | 1,43 | 0,97 | 1,06 | 1,16 | 1,25 | 1,34 | 1,42 | 1,51 | 1,06 | 1,15 | 1,25 | 1,34 | 1,42 | 1,51 | 1,59 |
| 24º | 1,22 | 1,34 | 1,44 | 1,56 | 1,63 | 1,79 | 1,00 | 1,32 | 1,44 | 1,54 | 1,66 | 1,78 | 1,89 | 2,00 | 1,43 | 1,55 | 1,65 | 1,77 | 1,89 | 2,00 | 2,11 |
| 25° | 1,61 | 1,66 | 1,63 | 1,98 | 2,12 | 2,26 | 2,40 | 1,66 | 1,31 | 1,96 | 2,11 | 2,25 | 2,39 | 2,52 | 1,80 | 1.94 | 2,09 | 2,24 | 2,39 | 2,52 | 2,66 |
| 26º | 1,87 | 2,05 | 2,22 | 2,40 | 2.56 | 2,71 | 2,87 | 2,02 | 2,20 | 2,37 | 2,54 | 2,70 | 2,55 | 3,01 | 2,13 | 2,36 | 2,53 | 2,71 | 2,86 | 3,02 | 3,17 |
| 27º | 2,21 | 2,42 | 2,60 | 2,80 | 3.00 | 3,18 | 3,35 | 2,39 | 2,59 | 2,73 | 2,93 | 3,17 | 3,35 | 3,52 | 2,53 | 2,78 | 2,97 | 3,17 | 3,36 | 3,54 | 3,71 |
| 28º | 2,56 | 2,30 | 3.02 | 3,25 | 3.47 | 3,67 | 3,39 | 2,75 | 2,69 | 3,22 | 3,44 | 3,66 | 3,96 | 4,07 | 2,97 | 3,21 | 3 44 | 3,66 | 3,88 | 4,09 | 4,30 |
| 29º | 2.93 | 3,19 | 3,43 | 3,66 | 3,91 | 4,14 | 4,37 | 3,16 | 3,41 | 3,65 | 3,39 | 4,13 | 4,36 | 4,59 | 3,40 | 3,66 | 3,89 | 4,13 | 4,38 | 4,61 | 4,82 |
| 30° | 3,31 | 3,57 | 3,86 | 4,15 | 4,41 | 4,66 | 4,92 | 3,55 | 3,81 | 4,10 | 4,38 | 4,66 | 4,90 | 5,16 | 3,82 | 4,08 | 4,37 | 4,65 | 4,93 | 5,17 | 5,42 |
Correcciones para referir la masa volúmica de los vinos dulces a 20°
| Temperaturas | MASAS VOLÚMICAS | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VINOS DE 19º | VINOS DE 21º | |||||||||||||
| 1,300 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,120 | 1,000 | 1.020 | 1,040 | 1,060 | 1,030 | 1,100 | 1,120 | |
| 10º | 3,14 | 3,48 | 3,83 | 4,17 | 4,48 | 4,78 | 5,07 | 3,50 | 3,84 | 4,19 | 4,52 | 4,83 | 5,12 | 5,41 |
| 11º | 2,87 | 3,18 | 3.49 | 3,73 | 4,06 | 4,32 | 4,57 | 3,18 | 3,49 | 3.80 | 4,09 | 4,34 | 4,63 | 4.88 |
| 12º | 2,58 | 2,36 | 3,13 | 3,39 | 3,65 | 3,88 | 4,10 | 2,86 | 3,13 | 3,41 | 3,67 | 3,92 | 4,15 | 4,37 |
| 13º | 2,31 | 2,55 | 2,77 | 2,99 | 3,20 | 3,41 | 3,61 | 2,56 | 2,79 | 3,01 | 3,23 | 3,44 | 3,65 | 3,85 |
| 14º | 2,03 | 2,23 | 2,43 | 2,61 | 2,80 | 2,96 | 3,13 | 2,23 | 2,43 | 2,03 | 2,81 | 3,00 | 3,16 | 3,33 |
| 15º | 1,89 | 1,85 | 2,02 | 2,18 | 2,33 | 2,48 | 2,62 | 1,86 | 2,03 | 2,19 | 2,35 | 2,50 | 2,65 | 2,80 |
| 16º | 1,38 | 1,53 | 1,65 | 1,78 | 1,90 | 2,02 | 2,13 | 1,51 | 1,65 | 1,78 | 1,91 | 2,03 | 2,15 | 2,26 |
| 17º | 1,06 | 1,16 | 1,26 | 1,35 | 1,44 | 1,53 | 1,62 | 1,15 | 1,25 | 1,35 | 1,45 | 1,54 | 1,63 | 1,71 |
| 18º | 0,73 | 0,79 | 0,85 | 0,92 | 0,98 | 1,03 | 1,09 | 0,79 | 0,85 | 0,92 | 0,98 | 1,05 | 1,10 | 1,15 |
| 19º | 0,38 | 0,41 | 0,44 | 0,43 | 0,51 | 0,52 | 0,56 | 0,41 | 0,44 | 0,47 | 0,51 | 0,54 | 0,57 | 0.59 |
| 20º | ||||||||||||||
| 21º | 0,37 | 0,41 | 0,44 | 0,47 | 0,50 | 0,53 | 0,56 | 0,41 | 0,44 | 0,47 | 0,51 | 0,54 | 0,57 | 0,59 |
| 22º | 0,75 | 0,81 | 0,87 | 0,93 | 0,99 | 1,04 | 1,10 | 0,81 | 0,88 | 0,94 | 1,00 | 1,06 | 1,10 | 1,17 |
| 23º | 1,15 | 1,30 | 1,34 | 1,43 | 1,51 | 1,60 | 1,68 | 1,25 | 1,34 | 1,44 | 1,63 | 1,61 | 1,70 | 1,78 |
| 24º | 1,55 | 1,67 | 1,77 | 1,89 | 2,00 | 2,11 | 2,23 | 1,68 | 1,80 | 1.90 | 2,02 | 2,13 | 2,25 | 2,36 |
| 25º | 1,95 | 2,09 | 2,24 | 2,39 | 2,53 | 2,67 | 2,71 | 2,11 | 2,25 | 2,40 | 2,55 | 2,69 | 2,83 | 2,97 |
| 26º | 2,38 | 2,54 | 2,71 | 2,89 | 3,04 | 3,20 | 3,35 | 2,55 | 2,73 | 2,90 | 3,07 | 3,22 | 3,38 | 3,54 |
| 27º | 2,79 | 2,09 | 3,18 | 3,38 | 3,57 | 3,75 | 3,92 | 3,01 | 3,20 | 3,40 | 3,59 | 3,78 | 3.96 | 4,13 |
| 28º | 3,20 | 3,44 | 3,66 | 3,89 | 4,11 | 4,32 | 4,53 | 3,46 | 3,69 | 3,93 | 4,15 | 4,36 | 4,58 | 4,77 |
| 29º | 3,65 | 3,92 | 4,15 | 4,40 | 4,64 | 4,87 | 5,08 | 3,95 | 4,20 | 4,43 | 4,68 | 4,92 | 5,15 | 5,36 |
| 30º | 4,11 | 4,27 | 4,65 | 4,84 | 5,22 | 5,46 | 5,71 | 4,42 | 4,68 | 4,97 | 5,25 | 5,53 | 5,77 | 6,02 |
TABLA 4(b).III
Cálculo de la densidad 20/20 a partir de la densidad 15/4
| Vinos secos (d, comprendida entre 0,97 y 1,03) | |||||||
|---|---|---|---|---|---|---|---|
|
Grados alcohólicos Correcciones c Grados alcohólicos Correcciones c Grados alcohólicos Correcciones c |
5 0,9 12 0,6 19 0,2 |
6 0,8 13 0,6 20 0,1 |
7 0,8 14 0,5 21 0,0 |
8 0,8 15 0,5 22 0,0 |
9 0,7 16 0,4 23 – 0,1 |
10 0,7 17 0,3 24 – 0,2 |
11 0,7 18 0,3 25 – 0,3 |
| Densidades | Vinos dulces | |||||||
|---|---|---|---|---|---|---|---|---|
| 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,200 | ||
| Grados alcohólicos |
10° 13° 15° 17° 19° 21° |
0,7 0,6 0,5 0,4 0,2 3,0 |
0,5 0,4 0,2 0,1 – 0,1 – 3,2 |
0,3 0,2 0,0 – 0,1 ‒ 0,3 – 0,4 |
0,1 0,0 – 0,2 ‒ 0,3 ‒ 0,5 ‒ 0,6 |
‒ 0,0 – 0,1 ‒ 0,3 ‒ 0,4 ‒ 0,6 ‒ 0,7 |
‒ 0,2 ‒ 0,3 ‒ 0,5 ‒ 0,6 ‒ 0,8 ‒ 0,9 |
‒ 0,3 ‒ 0,4 ‒ 0,6 ‒ 0,7 ‒ 0,9 – 1,0 |
TABLA 4(b).IV
Cálculo de la densidad 20/20 a partir de la densidad 15/15
| Vinos | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Grados alcohólicos Correcciones c Grados alcohólicos Correcciones c |
5 0,1 |
6 0,1 16 0,6 |
7 0,1 1,7 0,6 |
8 0,2 18 0,7 |
9 0,2 19 0,9 |
10 0,2 20 0,9 |
11 0,3 21 1,0 |
12 0,3 22 1,0 |
13 0,4 23 1,1 |
14 0,4 24 1,2 |
15 0,5 25 1,2 |
| Densidades | Vinos dulces | |||||||
|---|---|---|---|---|---|---|---|---|
| 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,200 | ||
| Grados alcohólicos |
10° 13° 15° 17° 19° 21° |
0,2 0,3 0,5 0,6 0,8 1,0 |
0,4 0,5 0,7 0,8 1,0 1,1 |
0,5 0,6 0,8 0,9 1,1 1,2 |
0,7 0,8 1,0 1,1 1,3 1,4 |
0,8 0,9 1,1 1,2 1,4 1,5 |
1,0 1,1 1,3 1,4 1,6 1,7 |
1,1 1,2 1,4 1,5 1,7 1,8 |
TABLA 4(b).V
Cálculo de la masa volúmica a 20º C (ρ20) a partir de la densidad 15/4
| Vinos | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Grados alcohólicos Correcciones c Grados alcohólicos Correcciones c |
5 0,9 |
6 1,0 16 1,4 |
7 1,0 17 1,5 |
3 1,0 18 1,5 |
9 1,1 19 1,8 |
10 1,1 20 1,7 |
11 1,1 21 1,8 |
12 1,2 22 1,8 |
13 1,2 23 1,9 |
14 1,3 24 2,0 |
15 1,3 25 2,1 |
| Densidades | Vinos dulces | |||||||
|---|---|---|---|---|---|---|---|---|
| 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,200 | ||
| Grados alcohólicos |
10° 13° 15° 17° 19° 21° |
1,1 1,2 1,3 1,4 1,6 1,8 |
1,3 1,4 1,6 1,7 1,9 2,0 |
1,5 1,6 1,8 1,9 2,1 2,2 |
1,7 1,8 2,0 2,1 2,3 2,4 |
1,8 1,9 2,1 2,2 2,4 2,5 |
2,0 2,1 2,3 2,4 2,6 2,7 |
2,1 2,2 2,4 2,5 2,7 2,8 |
TABLA 4(b).VI
Cálculo de la masa volúmica a 20º C (ρ20) a partir de la densidad 15/15
| Vinos | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Grados alcohólicos Correcciones c Grados alcohólicos Correcciones c |
5 1,8 |
6 1,9 16 2,3 |
7 1,9 17 2,4 |
8 1,9 18 2,4 |
9 2,0 19 2,5 |
10 2,0 20 2,6 |
11 2,0 21 2,7 |
12 2,1 22 2,7 |
13 2,1 23 2,3 |
14 2,2 24 2,9 |
15 2,2 25 3,0 |
| Densidades | Vinos dulces | |||||||
|---|---|---|---|---|---|---|---|---|
| 1,000 | 1,020 | 1,040 | 1,060 | 1,080 | 1,100 | 1,200 | ||
| Grados alcohólicos |
10° 13° 15° 17° 19° 21° |
2,0 2,1 2,2 2,3 2,5 2,7 |
2,2 2,3 2,5 2,6 2,8 2,9 |
2,4 2,5 2,7 2,8 3,0 3,1 |
2,6 2,7 2,9 3,0 3,2 3,3 |
2,7 2,8 3,0 3,1 3,3 3,4 |
2,9 3,1 3,3 3,4 3,6 3,7 |
3,1 3,2 3,4 3,5 3,7 3,8 |
5(a). TÍTULO ALCOHOMÉTRICO (1)
(Método picnométrico)
(1) Denominación adoptada para el grado alhólico por la Oficina Internacional del Vino (O. I. V.).
5(a).1 Principio.
El título alcohométrico es igual al número de litros de alcohol etílico contenidos en 100 l de vino, medidos ambos volúmenes a 20 °C, y se expresa en grados alcohólicos volumétricos, con una precisión de 0,1 °C.
Se determina por destilación simple de líquido alcalinizado y medida de la densidad del destilado por picnometría.
5(a).2 Material y aparatos.
5(a).2.1 Aparato de destilación.‒Consta de las siguientes partes:
5(a).2.1.1 Matraz de destilación de 1.000 ml con rodaje esmerilado.
5(a).2.1.2 Columna rectificadora de 20 cm de largo.
5(a).2.1.3 Disco metálico o de amianto con un orificio de 8 cm de diámetro.
5(a).2.1.4 Refrigerante de West de 40 cm de longitud, con circulación rápida de agua y dispuesto verticalmente.
5(a).2.2 Picnómetro.‒Como en 4(a).2.1, de vidrio pyrex.
5(a).2.3 Tara del picnómetro.‒Recipiente de vidrio del mismo volumen exterior que el picnómetro. Se llena casi por completo con una solución de cloruro de sodio, de densidad tal que la masa de esta tara sea igual a la masa del picnómetro lleno de un líquido de densidad 1,01. Este recipiente se cierra a la llama. Su capacidad exterior debe ser igual a la del picnómetro lleno, con una aproximación de 1 ml de diferencia.
5(a).2.4 Termostato y accesorios, descritos en 4(a).2.
5(a).2.5 Balanza analítica de 0,1 mg y 100 g de carga.
5(a).3 Reactivos.
5(a).3.1 Lechada de cal. Solución de 120 g de CaO en 1.000 mililitros de agua.
5(a).4 Procedimiento.
Para vinos jóvenes o espumosos, eliminar previamente el gas carbónico. Agitar 250 ml de vino en un matraz de 500 mililitros previamente siliconado el interior del matraz por tres gotas de solución al 1 por 100, de silicona soluble en agua, y después de bien seco, añadir el vino.
5(a).4.1 Destilación.‒Medir 250 ml de vino en matraz aforado con cuello de 12 mm de diámetro interior como máximo, y anotar la temperatura.
Introducir el vino en un matraz de destilación que contenga una docena de fragmentos de materia porosa (piedra pómez). Lavar el matraz cuatro veces con 5 ml de agua. Añadir 10 ml de la lechada de cal. La materia colorante del vino debe virar a la alcalinidad. En el caso vinos muy ácidos, puede comprobarse por toques externos el viraje de la fenolftaleína. Recoger el destilado en el mismo matraz de 250 ml, conteniendo unos 10 mililitros de agua pura, en la que debe sumergirse el pico del tubo afilado, prolongación del refrigerante. Destilar por lo menos 200 ml. Después agitar y llenar hasta volumen con agua, a la misma temperatura que se midió el vino inicialmente.
5(a).4.2 Tarado del picnómetro vacío.‒Poner en un platillo de la balanza la tara y en el otro el picnómetro vacío, limpio y seco, y a su lado, las masas necesarias p, para entre ambos equilibrar la balanza.
La masa equivalente a la de la tara será:
Masa del picnómetro + m + p
m = 0,0012 V.
m = masa en g del aire contenido en el picnómetro vacío.
V = volumen en ml del picnómetro.
0,0012 g/ml = masa volúmica del aire.
El volumen del picnómetro se considera, con suficiente aproximación, que es numéricamente igual a la masa del agua que llene el picnómetro.
Para calcular esta masa, colocar el picnómetro lleno de agua en el platillo de la balanza, si p es la masa que hay que colocar a su lado para equilibrar la de la tara, la masa m será: m = (p - p') · 0,0012.
Operar siempre a 20° exactamente para realizar el enrase, utilizando el termostato. Si se opera a t°, para conocer el volumen del picnómetro a 20°, hay que multiplicar (p ‒ p') por un factor F, que da la tabla 5 (a),I para picnómetro del vidrio pyrex.
Repetir estas operaciones de tara tres veces seguidas. Al cabo de un año, volver a comprobar la tara del picnómetro.
Con balanza monoplato, proceder análogamente. Primero poner la tara, hacer la lectura correspondiente en la escala de pesos y después poner el picnómetro vacío. Anotar los pesos p que hay que añadir hasta marcar la pesada que alcanzó la tara.
Tara = picnómetro + aire + p
5(a).4.3 Determinación picnométrica.‒Llenar el picnómetro lavado y seco con las precauciones expuestas en 4(a).3 con el destilado alcohólico a la temperatura ambiente, llevar después al termostato, agitar el líquido haciendo girar el picnómetro por dos o tres veces, hasta que la temperatura que marca el termómetro del picnómetro sea constante. Enrasar con el borde superior del tubo lateral, secar éste, colocarlo sobre el tapón receptor (para pequeños desbordes) y anotar la temperatura t° utilizando una lupa. Secar con el mismo cuidado el exterior del picnómetro y llevarlo al platillo de la balanza. Equilibrar la tara del picnómetro colocada en el otro platillo.
5(a).5 Cálculo.
Calcular el título alcohométrico expresado en grados alcohólicos volumétricos.
5(a).5.1 Cálculo de la masa volúmica aparente.

ρt = masa volúmica aparente.
m = masa del aire contenido en el picnómetro vacío.
p = diferencia de masa entre la tara y el picnómetro lleno con destilado alcohólico.
5(a).5.2 Cálculo de grado alcohométrico.
Calcular el grado alcohólico internacional O. I. V. a 20 °C utilizando la tabla 5 (a).1 de masas volúmicas a t ° corregidas del empuje del aire.
Buscar sobre la línea horizontal correspondiente a la cifra de la parte entera de la temperatura, t, la cifra de la masa volumen inmediatamente superior a pt. El grado alcohólico que encabeza la columna donde se encuentra esta cifra será la parte entera del grado alcohólico a 20 °C.
Determinar la masa volúmica ρ correspondiente a la parte entera de la temperatura, t utilizando la diferencia tabular que se lee por debajo de la masa volúmica inmediatamente superior a pt antes encontrada, y la cifra decimal t, ρ = ρt + (diferencia tabular × parte decimal de t) 10-5.
Hallar la parte decimal del grado alcohólico a 20°, buscando en las tablas sobre la línea horizontal que comienza con la cifra de los enteros de la temperatura, t, la masa volúmica ρ' que sea inmediatamente superior a ρ, y dividiendo la diferencia ρ' ‒ ρ por la diferencia tabular que está a la derecha de la cifra de la masa volúmica ρ.
5(a).6 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A2, 1-25. 1969.
2. P. Jaulmes, S. Brun: «Tables de correspondance entre les diverses tables alcoométriques». Société Pharmacie Montpellier, 26: 2, 111-141, 1966.
TABLA 5(a).I
Masas volúmicas aparentes de mezclas hidroalcohólicas (picnómetro Pyrex)
| Temperaturas | GRADOS ALCOHÓLICOS | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |||||||||||||
| 10º | 999 61 | 1 50 | 998 11 | 1 46 | 996 65 | 1 43 | 995 22 | 1 39 | 993 83 | 1 34 | 992 49 | 1 30 | 991 10 | 1 25 | 989 94 | 1 19 | 988 75 | 1 16 | 987 59 | 1 13 | 986 46 | 1 11 | 985 35 | 1 08 |
| 9 | 8 | 8 | 9 | 9 | 10 | 9 | 10 | 11 | 11 | 12 | 13 | |||||||||||||
| 11º | 999 52 | 1 49 | 998 03 | 1 46 | 996 57 | 1 44 | 995 13 | 1 39 | 993 74 | 1 35 | 992 39 | 1 29 | 991 10 | 1 26 | 989 84 | 1 20 | 988 64 | 1 16 | 987 48 | 1 14 | 986 34 | 1 21 | 985 22 | 1 09 |
| 10 | 10 | 10 | 10 | 10 | 10 | 11 | 11 | 12 | 12 | 13 | 15 | |||||||||||||
| 12º | 999 42 | 1 49 | 997 93 | 1 46 | 996 47 | 1 44 | 995 03 | 1 39 | 993 64 | 1 35 | 992 29 | 1 30 | 990 99 | 1 26 | 989 73 | 1 21 | 988 52 | 1 16 | 987 36 | 1 15 | 986 21 | 1 14 | 985 07 | 1 09 |
| 11 | 11 | 11 | 11 | 11 | 11 | 12 | 12 | 12 | 14 | 15 | 15 | |||||||||||||
| 13º | 999 31 | 1 49 | 997 82 | 1 46 | 996 36 | 1 44 | 994 92 | 1 39 | 993 53 | 1 35 | 992 18 | 1 31 | 990 87 | 1 26 | 989 61 | 1 21 | 988 40 | 1 18 | 987 22 | 1 16 | 986 06 | 1 14 | 984 92 | 1 11 |
| 12 | 12 | 13 | 12 | 13 | 13 | 13 | 13 | 14 | 15 | 15 | 16 | |||||||||||||
| 14° | 999 19 | 1 49 | 997 70 | 1 47 | 996 23 | 1 43 | 994 80 | 1 40 | 993 40 | 1 35 | 992 05 | 1 31 | 990 74 | 1 26 | 989 48 | 1 22 | 988 26 | 1 19 | 987 07 | 1 16 | 985 91 | 1 15 | 984 76 | 1 12 |
| 14 | 14 | 13 | 14 | 14 | 14 | 14 | 15 | 16 | 16 | 17 | 18 | |||||||||||||
| 15º | 999 05 | 1 49 | 997 56 | 1 46 | 996 10 | 1 44 | 994 66 | 1 40 | 993 26 | 1 35 | 991 91 | 1 31 | 990 60 | 1 27 | 989 33 | 1 23 | 988 10 | 1 19 | 986 91 | 1 17 | 985 74 | 1 16 | 984 58 | 1 13 |
| 14 | 14 | 15 | 14 | 14 | 15 | 16 | 16 | 16 | 17 | 18 | 10 | |||||||||||||
| 16º | 998 91 | 1 49 | 997 42 | 1 47 | 995 95 | 1 43 | 994 52 | 1 40 | 993 12 | 1 36 | 991 76 | 1 32 | 990 44 | 1 27 | 989 17 | 1 23 | 987 94 | 1 20 | 986 74 | 1 18 | 985 56 | 1 17 | 984 39 | 1 14 |
| 16 | 18 | 15 | 16 | 16 | 16 | 16 | 17 | 18 | 18 | 19 | 20 | |||||||||||||
| 17° | 998 75 | 1 49 | 997 26 | 1 46 | 995 80 | 1 44 | 994 36 | 1 40 | 992 96 | 1 36 | 991 60 | 1 32 | 990 28 | 1 28 | 989 00 | 1 24 | 987 76 | 1 20 | 986 56 | 1 19 | 935 37 | 1 18 | 984 19 | 1 15 |
| 17 | 17 | 17 | 17 | 18 | 18 | 18 | 18 | 18 | 19 | 20 | 21 | |||||||||||||
| 18º | 998 53 | 1 49 | 997 09 | 1 46 | 995 63 | 1 44 | 994 19 | 1 41 | 992 78 | 1 36 | 991 42 | 1 32 | 990 10 | 1 28 | 988 82 | 1 24 | 987 58 | 1 21 | 986 37 | 1 20 | 985 17 | 1 19 | 983 98 | 1 16 |
| 18 | 16 | 18 | 18 | 18 | 19 | 19 | 20 | 20 | 21 | 21 | 22 | |||||||||||||
| 19º | 998 40 | 1 49 | 996 91 | 1 46 | 995 45 | 1 44 | 994 01 | 1 41 | 992 60 | 1 37 | 991 23 | 1 32 | 989 91 | 1 29 | 988 62 | 1 24 | 987 38 | 1 22 | 986 16 | 1 20 | 984 96 | 1 20 | 983 76 | 1 17 |
| 19 | 19 | 20 | 20 | 19 | 19 | 20 | 20 | 21 | 22 | 23 | 23 | |||||||||||||
| 20º | 998 21 | 1 49 | 996 72 | 1 47 | 995 25 | 1 44 | 993 81 | 1 40 | 992 41 | 1 37 | 991 04 | 1 33 | 989 71 | 1 29 | 983 42 | 1 25 | 987 17 | 1 23 | 985 94 | 1 21 | 984 73 | 1 20 | 983 53 | 1 18 |
| 20 | 20 | 20 | 20 | 21 | 21 | 21 | 22 | 22 | 22 | 23 | 24 | |||||||||||||
| 21º | 999 01 | 1 49 | 996 52 | 1 47 | 995 05 | 1 44 | 993 81 | 1 41 | 992 20 | 1 37 | 990 83 | 1 33 | 989 50 | 1 30 | 988 20 | 1 25 | 986 95 | 1 23 | 985 72 | 1 22 | 984 50 | 1 21 | 933 29 | 1 19 |
| 21 | 21 | 21 | 21 | 21 | 22 | 22 | 22 | 23 | 24 | 22 | 26 | |||||||||||||
| 22º | 997 30 | 1 49 | 996 31 | 1 47 | 994 84 | 1 44 | 993 40 | 1 41 | 991 99 | 1 38 | 990 61 | 1 33 | 989 28 | 1 30 | 987 98 | 1 26 | 986 72 | 1 24 | 985 48 | 1 22 | 984 26 | 1 23 | 983 03 | 1 20 |
| 22 | 22 | 22 | 23 | 23 | 23 | 23 | 24 | 25 | 25 | 26 | 26 | |||||||||||||
| 23º | 957 58 | 1 49 | 995 09 | 1 47 | 994 62 | 1 45 | 993 17 | 1 41 | 991 76 | 1 38 | 990 38 | 1 33 | 989 05 | 1 31 | 987 74 | 1 27 | 986 47 | 1 24 | 985 23 | 1 23 | 984 00 | 1 23 | 932 77 | 1 21 |
| 24 | 24 | 24 | 23 | 24 | 24 | 25 | 25 | 25 | 26 | 27 | 27 | |||||||||||||
| 24º | 997 34 | 1 49 | 995 85 | 1 47 | 994 38 | 1 44 | 992 94 | 1 42 | 991 52 | 1 38 | 990 14 | 1 34 | 988 80 | 1 31 | 987 49 | 1 27 | 986 22 | 1 25 | 984 97 | 1 24 | 983 73 | 1 23 | 982 50 | 1 22 |
| 24 | 24 | 24 | 25 | 25 | 25 | 25 | 25 | 26 | 27 | 27 | 29 | |||||||||||||
| 25º | 997 10 | 1 49 | 995 61 | 1 47 | 994 14 | 1 45 | 992 69 | 1 42 | 991 27 | 1 38 | 989 89 | 1 34 | 988 55 | 1 31 | 987 24 | 1 28 | 985 96 | 1 26 | 984 70 | 1 24 | 983 46 | 1 25 | 982 21 | 1 23 |
| 25 | 25 | 25 | 25 | 25 | 26 | 26 | 27 | 27 | 27 | 29 | 29 | |||||||||||||
| 26º | 996 85 | 1 49 | 995 36 | 1 47 | 993 89 | 1 45 | 992 44 | 1 42 | 991 02 | 1 39 | 989 63 | 1 34 | 988 29 | 1 32 | 986 97 | 1 23 | 985 69 | 1 26 | 984 43 | 1 26 | 983 17 | 1 25 | 981 92 | 1 24 |
| 26 | 26 | 27 | 27 | 27 | 27 | 28 | 28 | 29 | 29 | 29 | 31 | |||||||||||||
| 27° | 996 59 | 1 49 | 995 10 | 1 48 | 993 82 | 1 45 | 992 17 | 1 42 | 990 75 | 1 39 | 989 36 | 1 35 | 988 01 | 1 32 | 986 69 | 1 29 | 985 40 | 1 26 | 984 14 | 1 26 | 982 88 | 1 27 | 981 61 | 1 24 |
| 27 | 26 | 27 | 27 | 28 | 28 | 28 | 29 | 29 | 30 | 31 | 31 | |||||||||||||
| 28° | 996 32 | 1 50 | 994 82 | 1 47 | 993 35 | 1 45 | 991 90 | 1 43 | 990 47 | 1 39 | 989 08 | 1 35 | 987 73 | 1 33 | 986 40 | 1 29 | 985 11 | 1 27 | 983 84 | 1 27 | 982 57 | 1 27 | 981 30 | 1 26 |
| 28 | 28 | 28 | 28 | 28 | 28 | 29 | 29 | 30 | 31 | 31 | 32 | |||||||||||||
| 29º | 996 04 | 1 50 | 994 54 | 1 47 | 993 07 | 1 46 | 991 61 | 1 42 | 990 19 | 1 39 | 988 30 | 1 36 | 987 44 | 1 33 | 986 11 | 1 30 | 984 81 | 1 28 | 983 53 | 1 27 | 982 26 | 1 28 | 980 98 | 1 27 |
| 29 | 20 | 29 | 29 | 30 | 30 | 30 | 31 | 31 | 31 | 32 | 33 | |||||||||||||
| 30º | 995 75 | 1 50 | 994 25 | 1 47 | 992 78 | 1 46 | 991 32 | 1 43 | 989 69 | 1 39 | 988 50 | 1 36 | 987 14 | 1 34 | 985 80 | 1 30 | 984 50 | 1 28 | 983 22 | 1 28 | 981 94 | 1 29 | 980 65 | 1 28 |
| Temperaturas | GRADOS ALCOHÓLICOS | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | |||||||||||||
| 10º | 986 46 | 1 11 | 935 35 | 1 08 | 984 27 | 1 04 | 983 23 | 1 02 | 982 21 | 99 | 961 22 | 98 | 980 24 | 94 | 979 30 | 91 | 978 36 | 91 | 977 45 | 89 | 976 56 | 90 | 975 66 | 91 |
| 12 | 13 | 14 | 15 | 16 | 18 | 19 | 21 | 22 | 24 | 25 | 27 | |||||||||||||
| 11º | 986 34 | 1 12 | 935 22 | 1 09 | 984 13 | 1 05 | 983 08 | 1 03 | 982 05 | 1 01 | 981 04 | 98 | 980 05 | 96 | 979 09 | 95 | 978 14 | 93 | 977 21 | 90 | 976 31 | 92 | 975 39 | 93 |
| 13 | 15 | 15 | 17 | 18 | 19 | 20 | 22 | 23 | 25 | 27 | 28 | |||||||||||||
| 12º | 986 21 | 1 14 | 935 07 | 1 09 | 983 98 | 1 07 | 982 91 | 1 04 | 981 37 | 1 02 | 980 85 | 1 00 | 970 85 | 93 | 978 87 | 96 | 977 91 | 95 | 976 96 | 92 | 976 04 | 93 | 975 11 | 95 |
| 15 | 15 | 17 | 17 | 19 | 20 | 22 | 23 | 25 | 26 | 27 | 30 | |||||||||||||
| 13º | 985 06 | 1 14 | 984 92 | 1 11 | 983 81 | 1 07 | 932 74 | 1 06 | 981 63 | 1 03 | 980 65 | 1 02 | 979 63 | 99 | 978 64 | 98 | 977 66 | 96 | 976 70 | 93 | 975 77 | 96 | 974 31 | 96 |
| 15 | 16 | 17 | 19 | 19 | 21 | 23 | 24 | 25 | 26 | 29 | 30 | |||||||||||||
| 14º | 985 91 | 1 15 | 984 76 | 1 12 | 983 64 | 1 09 | 932 55 | 1 06 | 981 49 | 1 05 | 980 44 | 1 03 | 979 41 | 1 01 | 978 40 | 99 | 977 41 | 97 | 976 44 | 96 | 975 48 | 97 | 974 51 | 98 |
| 17 | 18 | 19 | 20 | 22 | 22 | 24 | 25 | 26 | 28 | 29 | 31 | |||||||||||||
| 15º | 985 74 | 1 16 | 984 52 | 1 13 | 983 45 | 1 10 | 982 35 | 1 03 | 981 27 | 1 05 | 930 22 | 1 05 | 979 17 | 1 02 | 973 15 | 1 00 | 977 15 | 97 | 976 16 | 97 | 975 19 | 99 | 974 20 | 1 00 |
| 18 | 19 | 20 | 21 | 22 | 24 | 24 | 26 | 28 | 29 | 31 | 32 | |||||||||||||
| 16º | 935 56 | 1 17 | 984 39 | 1 14 | 983 25 | 1 11 | 982 14 | 1 09 | 981 05 | 1 07 | 979 98 | 1 05 | 978 93 | 1 04 | 977 80 | 1 02 | 976 87 | 1 00 | 975 87 | 99 | 974 88 | 1 00 | 973 88 | 1 01 |
| 19 | 20 | 21 | 22 | 23 | 24 | 26 | 27 | 28 | 30 | 31 | 33 | |||||||||||||
| 17º | 935 37 | 1 19 | 984 10 | 1 15 | 983 04 | 1 12 | 981 92 | 1 10 | 980 82 | 1 08 | 979 74 | 1 07 | 978 67 | 1 05 | 977 62 | 1 03 | 976 59 | 1 02 | 975 57 | 1 00 | 974 57 | 1 02 | 973 55 | 1 03 |
| 20 | 21 | 22 | 23 | 24 | 26 | 27 | 28 | 30 | 31 | 32 | 34 | |||||||||||||
| 18º | 985 17 | 1 19 | 983 91 | 1 16 | 982 32 | 1 13 | 981 45 | 1 12 | 980 58 | 1 10 | 979 48 | 1 08 | 978 40 | 1 06 | 977 34 | 1 05 | 976 29 | 1 03 | 975 26 | 1 01 | 974 25 | 1 04 | 973 21 | 1 05 |
| 21 | 22 | 23 | 24 | 25 | 26 | 25 | 29 | 30 | 32 | 34 | 35 | |||||||||||||
| 19º | 984 96 | 1 20 | 983 71 | 1 17 | 982 59 | 1 14 | 981 45 | 1 12 | 980 33 | 1 11 | 979 22 | 1 10 | 973 12 | 1 07 | 977 05 | 1 06 | 975 99 | 1 05 | 974 94 | 1 03 | 973 91 | 1 05 | 972 86 | 1 03 |
| 22 | 23 | 24 | 26 | 27 | 28 | 28 | 30 | 32 | 33 | 34 | 36 | |||||||||||||
| 20º | 984 73 | 1 20 | 983 53 | 1 13 | 982 35 | 1 16 | 981 19 | 1 13 | 980 06 | 1 12 | 978 94 | 1 10 | 977 84 | 1 09 | 976 75 | 1 08 | 975 67 | 1 06 | 974 61 | 1 04 | 973 57 | 1 07 | 972 50 | 1 08 |
| 23 | 24 | 25 | 26 | 27 | 28 | 30 | 31 | 32 | 34 | 35 | 36 | |||||||||||||
| 21º | 984 50 | 1 21 | 983 29 | 1 19 | 982 10 | 1 17 | 980 93 | 1 14 | 979 79 | 1 13 | 978 66 | 1 12 | 977 54 | 1 10 | 976 44 | 1 09 | 975 35 | 1 08 | 974 27 | 1 05 | 973 22 | 1 08 | 972 14 | 1 10 |
| 24 | 26 | 27 | 27 | 29 | 30 | 31 | 32 | 33 | 34 | 36 | 38 | |||||||||||||
| 22º | 984 26 | 1 23 | 933 03 | 1 20 | 981 83 | 1 17 | 980 66 | 1 16 | 979 50 | 1 14 | 978 36 | 1 13 | 977 23 | 1 11 | 976 12 | 1 10 | 975 02 | 1 09 | 973 93 | 1 07 | 972 86 | 1 10 | 971 76 | 1 11 |
| 26 | 26 | 27 | 28 | 29 | 30 | 31 | 33 | 34 | 36 | 37 | 38 | |||||||||||||
| 23º | 984 00 | 1 23 | 982 77 | 1 21 | 981 56 | 1 18 | 980 38 | 1 17 | 979 21 | 1 15 | 978 06 | 1 14 | 976 92 | 1 13 | 975 79 | 1 11 | 974 68 | 1 11 | 973 57 | 1 08 | 972 40 | 1 11 | 971 38 | 1 13 |
| 27 | 27 | 28 | 30 | 30 | 32 | 33 | 34 | 36 | 36 | 38 | 40 | |||||||||||||
| 24º | 983 73 | 1 23 | 982 50 | 1 22 | 981 28 | 1 20 | 980 02 | 1 17 | 978 91 | 1 17 | 977 74 | 1 15 | 976 59 | 1 14 | 975 45 | 1 13 | 974 32 | 1 11 | 973 21 | 1 10 | 972 11 | 1 13 | 970 98 | 1 14 |
| 27 | 28 | 30 | 30 | 32 | 32 | 34 | 34 | 36 | 38 | 39 | 40 | |||||||||||||
| 25º | 983 46 | 1 25 | 982 21 | 1 23 | 980 89 | 120 | 979 78 | 1 19 | 973 59 | 1 17 | 977 42 | 1 17 | 976 25 | 1 14 | 975 11 | 1 15 | 973 96 | 1 13 | 972 83 | 1 11 | 971 72 | 1 14 | 9,0 58 | 1 15 |
| 29 | 29 | 30 | 31 | 32 | 34 | 34 | 36 | 37 | 38 | 40 | 41 | |||||||||||||
| 26º | 983 17 | 1 25 | 981 92 | 1 24 | 980 88 | 1 21 | 979 47 | 1 20 | 978 27 | 1 19 | 977 05 | 1 17 | 975 91 | 1 16 | 974 75 | 1 16 | 973 59 | 1 14 | 972 45 | 1 13 | 971 22 | 1 15 | 970 17 | 1 17 |
| 29 | 31 | 31 | 33 | 33 | 34 | 36 | 37 | 33 | 99 | 40 | 42 | |||||||||||||
| 27º | 982 88 | 1 27 | 981 61 | 1 24 | 980 37 | 1 23 | 979 14 | 1 20 | 977 94 | 1 20 | 976 74 | 1 19 | 975 55 | 1 17 | 974 38 | 1 17 | 973 21 | 1 15 | 972 06 | 1 14 | 970 92 | 1 17 | 969 15 | 1 18 |
| 31 | 31 | 33 | 33 | 35 | 35 | 36 | 37 | 38 | 40 | 41 | 42 | |||||||||||||
| 28º | 982 57 | 1 27 | 981 20 | 1 26 | 980 04 | 1 23 | 978 81 | 1 22 | 977 59 | 1 20 | 976 39 | 1 20 | 975 19 | 1 18 | 974 01 | 1 18 | 972 83 | 1 17 | 971 66 | 1 15 | 970 51 | 1 18 | 969 33 | 1 20 |
| 31 | 32 | 33 | 34 | 35 | 36 | 37 | 39 | 40 | 41 | 43 | 44 | |||||||||||||
| 29º | 982 26 | 1 23 | 980 93 | 1 27 | 979 71 | 1 24 | 978 47 | 1 23 | 977 24 | 1 21 | 976 03 | 1 21 | 974 82 | 1 20 | 973 62 | 1 19 | 972 43 | 1 18 | 971 25 | 1 17 | 970 08 | 1 19 | 968 89 | 1 21 |
| 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 42 | 42 | 44 | |||||||||||||
| 30º | 981 94 | 1 29 | 980 55 | 1 28 | 979 37 | 1 25 | 978 12 | 1 24 | 976 88 | 1 22 | 975 66 | 1 22 | 974 44 | 1 21 | 973 23 | 1 20 | 972 03 | 1 20 | 970 83 | 1 17 | 969 66 | 1 21 | 968 45 | 1 23 |
| Temperaturas | GRADOS ALCOHÓLICOS | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | ||||||||||||
| 10º | 977 45 | 89 | 976 56 | 90 | 975 66 | 91 | 974 75 | 91 | 973 84 | 94 | 972 90 | 94 | 971 96 | 94 | 971 02 | 97 | 970 05 | 99 | 969 06 | 1 01 | 966 05 | 1 03 | 937 02 |
| 24 | 25 | 27 | 29 | 32 | 32 | 35 | 37 | 39 | 41 | 43 | 45 | ||||||||||||
| 11º | 977 21 | 90 | 976 31 | 92 | 975 39 | 93 | 974 46 | 94 | 973 52 | 94 | 972 58 | 97 | 971 01 | 96 | 970 65 | 99 | 969 66 | 1 01 | 968 65 | 1 03 | 967 62 | 1 05 | 966 57 |
| 25 | 27 | 28 | 30 | 31 | 34 | 35 | 38 | 40 | 42 | 44 | 46 | ||||||||||||
| 12º | 976 96 | 92 | 976 04 | 93 | 975 11 | 95 | 974 16 | 95 | 973 21 | 97 | 972 24 | 98 | 971 26 | 99 | 970 27 | 1 01 | 960 26 | 1 03 | 968 23 | 1 05 | 967 18 | 1 07 | 963 11 |
| 26 | 27 | 30 | 31 | 33 | 35 | 37 | 38 | 40 | 42 | 44 | 46 | ||||||||||||
| 13º | 976 70 | 93 | 975 77 | 96 | 974 81 | 96 | 973 85 | 97 | 972 88 | 99 | 971 39 | 1 00 | 970 89 | 1 00 | 969 89 | 1 03 | 960 66 | 1 05 | 967 81 | 1 07 | 966 74 | 1 09 | 965 65 |
| 26 | 29 | 30 | 32 | 34 | 35 | 37 | 40 | 42 | 44 | 46 | 47 | ||||||||||||
| 14º | 976 44 | 96 | 975 48 | 97 | 974 51 | 98 | 973 53 | 99 | 972 54 | 1 00 | 971 54 | 1 02 | 970 52 | 1 03 | 969 49 | 1 05 | 968 44 | 1 07 | 967 07 | 1 09 | 966 23 | 1 10 | 965 18 |
| 23 | 29 | 31 | 33 | 35 | 37 | 39 | 40 | 42 | 44 | 46 | 48 | ||||||||||||
| 15º | 976 16 | 97 | 975 19 | 99 | 974 20 | 1 00 | 973 20 | 1 01 | 972 19 | 1 02 | 971 17 | 1 04 | 970 13 | 1 04 | 969 09 | 1 07 | 968 02 | 1 09 | 966 93 | 1 11 | 965 82 | 1 12 | 964 70 |
| 29 | 31 | 32 | 33 | 35 | 37 | 39 | 41 | 43 | 44 | 46 | 48 | ||||||||||||
| 16º | 975 87 | 99 | 974 83 | 1 00 | 973 88 | 1 01 | 972 87 | 1 03 | 971 84 | 1 04 | 970 80 | 1 06 | 969 74 | 1 06 | 968 68 | 1 00 | 967 59 | 1 10 | 960 49 | 1 13 | 965 36 | 1 14 | 964 22 |
| 30 | 31 | 33 | 35 | 36 | 38 | 40 | 42 | 43 | 46 | 47 | 49 | ||||||||||||
| 17° | 975 57 | 1 00 | 974 57 | 1 02 | 973 55 | 1 03 | 972 52 | 1 04 | 971 48 | 1 06 | 970 42 | 1 08 | 969 34 | 1 08 | 968 26 | 1 10 | 967 16 | 1 13 | 966 03 | 1 14 | 964 89 | 1 16 | 963 73 |
| 31 | 32 | 34 | 36 | 38 | 40 | 41 | 42 | 45 | 46 | 48 | 50 | ||||||||||||
| 18º | 975 26 | 1 01 | 974 25 | 1 04 | 973 21 | 1 05 | 972 16 | 1 06 | 971 10 | 1 08 | 970 02 | 1 09 | 969 63 | 1 09 | 967 84 | 1 13 | 966 71 | 1 14 | 965 67 | 1 16 | 964 41 | 1 18 | 963 23 |
| 32 | 34 | 35 | 36 | 38 | 39 | 41 | 44 | 45 | 47 | 48 | 50 | ||||||||||||
| 19º | 974 94 | 1 03 | 973 91 | 1 05 | 972 86 | 1 06 | 971 80 | 1 08 | 970 72 | 1 09 | 969 63 | 1 11 | 968 52 | 1 12 | 967 40 | 1 14 | 966 26 | 1 16 | 965 10 | 1 18 | 963 92 | 1 19 | 962 73 |
| 33 | 34 | 36 | 38 | 39 | 41 | 43 | 44 | 46 | 48 | 49 | 51 | ||||||||||||
| 20º | 974 61 | 1 04 | 973 57 | 1 07 | 972 59 | 1 08 | 971 42 | 1 09 | 970 33 | 1 11 | 969 22 | 1 13 | 968 09 | 1 13 | 966 96 | 1 16 | 965 80 | 1 18 | 964 62 | 1 19 | 963 43 | 1 21 | 962 22 |
| 34 | 35 | 38 | 38 | 40 | 41 | 43 | 45 | 46 | 48 | 50 | 52 | ||||||||||||
| 21º | 974 27 | 1 05 | 973 22 | 1 08 | 972 14 | 1 10 | 971 04 | 1 11 | 969 93 | 1 12 | 968 81 | 1 15 | 967 66 | 1 15 | 966 51 | 1 17 | 965 34 | 1 20 | 964 14 | 1 21 | 962 00 | 1 23 | 961 70 |
| 34 | 36 | 38 | 39 | 41 | 43 | 44 | 45 | 47 | 49 | 50 | 52 | ||||||||||||
| 22° | 973 93 | 1 07 | 972 86 | 1 10 | 971 76 | 1 11 | 970 65 | 1 13 | 969 52 | 1 14 | 965 38 | 1 16 | 967 22 | 1 16 | 966 06 | 1 19 | 964 87 | 1 22 | 963 65 | 1 22 | 962 43 | 1 25 | 961 18 |
| 36 | 37 | 38 | 40 | 41 | 43 | 44 | 47 | 48 | 49 | 52 | 53 | ||||||||||||
| 23º | 973 57 | 1 08 | 972 49 | 1 11 | 971 38 | 1 13 | 970 25 | 1 14 | 969 11 | 1 16 | 967 95 | 1 17 | 966 78 | 1 19 | 965 59 | 1 20 | 964 39 | 1 23 | 963 16 | 1 25 | 961 91 | 1 26 | 960 66 |
| 38 | 38 | 40 | 41 | 42 | 44 | 46 | 47 | 49 | 51 | 52 | 53 | ||||||||||||
| 24º | 973 21 | 1 10 | 972 11 | 1 13 | 970 96 | 1 14 | 969 84 | 1 15 | 968 69 | 1 18 | 967 51 | 1 10 | 966 32 | 1 20 | 965 12 | 1 22 | 963 90 | 1 25 | 962 65 | 1 26 | 961 39 | 1 27 | 960 12 |
| 38 | 39 | 40 | 41 | 44 | 45 | 65 | 48 | 50 | 51 | 52 | 56 | ||||||||||||
| 25º | 972 83 | 1 11 | 371 72 | 1 14 | 970 58 | 1 15 | 969 43 | 1 18 | 966 25 | 1 19 | 967 06 | 1 20 | 965 86 | 1 22 | 964 54 | 1 24 | 963 40 | 1 26 | 952 14 | 1 27 | 960 87 | 1 30 | 959 67 |
| 38 | 40 | 41 | 43 | 44 | 45 | 47 | 48 | 50 | 51 | 53 | 54 | ||||||||||||
| 26º | 972 45 | 1 13 | 971 32 | 1 15 | 970 17 | 1 17 | 969 00 | 1 19 | 967 81 | 1 20 | 966 61 | 1 22 | 985 39 | 1 23 | 964 16 | 1 26 | 962 90 | 1 27 | 951 63 | 1 29 | 960 34 | 1 31 | 959 03 |
| 39 | 40 | 42 | 44 | 44 | 46 | 48 | 48 | 50 | 52 | 54 | 55 | ||||||||||||
| 27º | 972 06 | 1 14 | 970 92 | 1 17 | 969 75 | 1 18 | 968 57 | 1 20 | 967 37 | 1 22 | 966 15 | 1 24 | 964 91 | 1 24 | 963 67 | 1 27 | 962 40 | 1 29 | 961 11 | 1 31 | 959 80 | 1 32 | 959 58 |
| 40 | 41 | 42 | 44 | 46 | 47 | 48 | 50 | 52 | 53 | 54 | 56 | ||||||||||||
| 28º | 371 66 | 1 15 | 970 51 | 1 18 | 969 33 | 1 20 | 968 13 | 1 22 | 966 91 | 1 23 | 965 68 | 1 25 | 964 43 | 1 26 | 963 17 | 1 23 | 961 88 | 1 30 | 960 58 | 1 32 | 959 20 | 1 34 | 957 92 |
| 41 | 43 | 44 | 45 | 46 | 48 | 49 | 51 | 52 | 54 | 55 | 57 | ||||||||||||
| 29º | 971 25 | 1 17 | 970 03 | 1 19 | 963 89 | 1 21 | 967 63 | 1 23 | 966 46 | 1 25 | 965 20 | 1 26 | 963 94 | 1 28 | 962 66 | 1 30 | 961 36 | 1 32 | 960 04 | 1 33 | 953 71 | 1 36 | 957 35 |
| 42 | 42 | 44 | 45 | 47 | 48 | 50 | 51 | 53 | 54 | 56 | 57 | ||||||||||||
| 30º | 970 83 | 1 17 | 969 66 | 1 21 | 968 45 | 1 23 | 967 22 | 1 24 | 965 98 | 1 26 | 964 72 | 1 28 | 983 44 | 1 29 | 962 15 | 1 32 | 960 83 | 1 33 | 959 50 | 1 35 | 958 15 | 1 37 | 956 78 |
5(b). TÍTULO ALCOHOMÉTRICO
(Método areométrico)
5(b).1 Principio.
Como en 5(a).1.
5(b).2 Material y aparatos.
5(b).2.1 Aparatos de destilación.‒Como en 5(a).2.1.
5(b).2.2 Aparatos para areometría.
5(b).2.2.1 Alcohómetro.
5(b).2.2.2 Termómetro.
5(b).2.2.3 Probeta.
5(b).3 Reactivos.
Como en 5(a).3.
5(b).4 Procedimiento.
Las generalidades como en 5(a).4.
5(b).4.1 Destilación.‒Como en 5(a).4.1.
5(b).4.2 Determinación areométrica.‒Como en 4(b).3, teniendo en cuenta que se utiliza destilado en vez de vino, y que el tallo del areómetro viene graduado en grados alcohólicos aparentes. Hacer al menos tres lecturas del grado alcohólico aparente, sirviéndose si es preciso de una lupa.
5(b).5 Cálculo.
Calcular el grado alcohólico internacional O. I. V. a 20 °C, utilizando la tabla 5(b),I, añadiendo o restando al grado alcohólico aparente a t° la corrección correspondiente.
5(b).6 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A2, 1-25. 1969.
2. P. Jaulmes, S, Brun: «Tables de correspondance entre les diverses tables alcoométriques». Société Pharmacie Montpellier, 26: 2, 111-141, 1966.
TABLA 5(b).I
Correcciones a efectuar, sobre el grado alcohólico aparente para corregir la acción de la temperatura
| Temperaturas | Grados alcohólicos aparentes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | ||
| 10º | Sumar | 0,84 | 0,87 | 0,88 | 0,92 | 0,96 | 1,01 | 1,08 | 1,16 | 1,24 | 1,34 | 1,44 | 1,58 | 1,73 | 1,89 | 2,06 | 2,24 | 2,43 | 2,63 |
| 11° | 0,80 | 0,81 | 0,83 | 0,86 | 0,90 | 0,96 | 1,01 | 1,08 | 1,16 | 1,25 | 1,34 | 1,46 | 1,59 | 1,73 | 1,88 | 2,04 | 2,21 | 2,38 | |
| 12° | 0,74 | 0,76 | 0,78 | 0,80 | 0,84 | 0,88 | 0,94 | 1,00 | 1,07 | 1,15 | 1,23 | 1,32 | 1,43 | 1,55 | 1,68 | 1,82 | 1,97 | 2,13 | |
| 13° | 0,67 | 0,69 | 0,70 | 0,73 | 0,76 | 0,80 | 0,86 | 0,90 | 0,95 | 1,02 | 1,10 | 1,18 | 1,28 | 1,38 | 1,48 | 1,60 | 1,72 | 1,85 | |
| 14° | 0,60 | 0,61 | 0,62 | 0,65 | 0,68 | 0,71 | 0,75 | 0,79 | 0,85 | 0,90 | 0,96 | 1,03 | 1,11 | 1,19 | 1,28 | 1,39 | 1,49 | 1,59 | |
| 15° | 0,51 | 0,52 | 0,54 | 0,56 | 0,58 | 0,61 | 0,64 | 0,68 | 0,72 | 0,77 | 0,82 | 0,87 | 0,94 | 1,01 | 1,09 | 1,17 | 1,26 | 1,34 | |
| 16° | 0,42 | 0,43 | 0,45 | 0,46 | 0,48 | 0,50 | 0,53 | 0,55 | 0,58 | 0,62 | 0,66 | 0,71 | 0,76 | 0,82 | 0,88 | 0,94 | 1,01 | 1,08 | |
| 17° | 0,32 | 0,33 | 0,34 | 0,35 | 0,37 | 0,39 | 0,41 | 0,43 | 0,46 | 0,48 | 0,51 | 0,55 | 0,58 | 0,63 | 0,67 | 0,71 | 0,76 | 0,81 | |
| 18° | 0,23 | 0,24 | 0,24 | 0,25 | 0,26 | 0,27 | 0,28 | 0,30 | 0,31 | 0,33 | 0,34 | 0,36 | 0,39 | 0,42 | 0,45 | 0,47 | 0,50 | 0,53 | |
| 19° | 0,11 | 0,12 | 0,12 | 0,13 | 0,13 | 0,13 | 0,14 | 0,15 | 0,16 | 0,16 | 0,17 | 0,18 | 0,20 | 0,21 | 0,23 | 0,24 | 0,26 | 0,28 | |
| 20° | |||||||||||||||||||
| 21° | Restar | 0,13 | 0,13 | 0,13 | 0,14 | 0,14 | 0,15 | 0,15 | 0,16 | 0,17 | 0,18 | 0,19 | 0,20 | 0,21 | 0,23 | 0,24 | 0,26 | 0,28 | |
| 22° | 0,26 | 0,27 | 0,28 | 0,28 | 0,29 | 0,30 | 0,32 | 0,34 | 0,35 | 0,37 | 0,40 | 0,42 | 0,44 | 0,46 | 0,48 | 0,51 | 0,54 | ||
| 23° | 0,41 | 0,42 | 0,43 | 0,44 | 0,45 | 0,47 | 0,49 | 0,51 | 0,54 | 0,56 | 0,60 | 0,63 | 0,67 | 0,71 | 0,74 | 0,78 | 0,82 | ||
| 24° | 0,56 | 0,57 | 0,58 | 0,59 | 0,61 | 0,64 | 0,67 | 0,70 | 0,73 | 0,77 | 0,81 | 0,85 | 0,89 | 0,94 | 0,99 | 1,04 | 1,09 | ||
| 25° | 0,71 | 0,72 | 0,73 | 0,75 | 0,78 | 0,81 | 0,84 | 0,89 | 0,93 | 0,98 | 1,02 | 1,07 | 1,12 | 1,18 | 1,24 | 1,32 | 1,38 | ||
| 26° | 0,86 | 0,88 | 0,91 | 0,93 | 0,96 | 1,00 | 1,04 | 1,09 | 1,14 | 1,19 | 1,24 | 1,30 | 1,36 | 1,43 | 1,51 | 1,57 | 1,65 | ||
| 27° | 1,05 | 1,08 | 1,10 | 1,14 | 1,18 | 1,23 | 1,28 | 1,34 | 1,40 | 1,46 | 1,53 | 1,60 | 1,68 | 1,76 | 1,85 | 1,93 | |||
| 28° | 1,22 | 1,25 | 1,28 | 1,33 | 1,38 | 1,44 | 1,50 | 1,56 | 1,62 | 1,68 | 1,75 | 1,83 | 1,92 | 2,02 | 2,11 | 2,21 | |||
| 29° | 1,40 | 1,44 | 1,48 | 1,52 | 1,58 | 1,64 | 1,71 | 1,78 | 1,85 | 1,92 | 2,00 | 2,08 | 2,17 | 2,28 | 2,39 | 2,50 | |||
| 30º | 1,59 | 1,62 | 1,66 | 1,71 | 1,77 | 1,85 | 1,93 | 2,00 | 2,07 | 2,15 | 2,23 | 2,33 | 2,45 | 2,55 | 2,67 | 2,79 | |||
| Temperaturas | Grados alcohólicos aparentes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | ||
| 10° | Sumar | 1,89 | 2,06 | 2,24 | 2,43 | 2,63 | 2,84 | 3,01 | 3,15 | 3,29 | 3,44 | 3,58 | 3,70 | 3,82 | 3,93 | 4,04 | 4,14 | 4,24 | 4,33 |
| 11º | 1,73 | 1,88 | 2,04 | 2,21 | 2,38 | 2,56 | 2,71 | 2,84 | 2,95 | 3,08 | 3,21 | 3,32 | 3,42 | 3,52 | 3,61 | 3,69 | 3,78 | 3,86 | |
| 12° | 1,55 | 1,68 | 1,82 | 1,97 | 2,13 | 2,27 | 2,40 | 2,51 | 2,63 | 2,73 | 2,84 | 2,93 | 3,02 | 3,10 | 3,18 | 3,25 | 3,32 | 3,39 | |
| 13º | 1,38 | 1,48 | 1,60 | 1,72 | 1,85 | 1,99 | 2,11 | 2,20 | 2,29 | 2,38 | 2,47 | 2,57 | 2,64 | 2,71 | 2,78 | 2,84 | 2,89 | 2,94 | |
| 14° | 1,19 | 1,28 | 1,39 | 1,49 | 1,59 | 1,71 | 1,81 | 1,89 | 1,96 | 2,03 | 2,11 | 2,19 | 2,25 | 2,30 | 2,36 | 2,42 | 2,47 | 2,52 | |
| 15° | 1,01 | 1,09 | 1,17 | 1,26 | 1,34 | 1,44 | 1,51 | 1,58 | 1,64 | 1,71 | 1,77 | 1,82 | 1,87 | 1,91 | 1,96 | 2,01 | 2,05 | 2,09 | |
| 16° | 0,82 | 0,88 | 0,94 | 1,01 | 1,08 | 1,15 | 1,20 | 1,26 | 1,31 | 1,36 | 1,40 | 1,45 | 1,49 | 1,53 | 1,56 | 1,59 | 1,62 | 1,64 | |
| 17° | 0,63 | 0,67 | 0,71 | 0,76 | 0,81 | 0,85 | 0,90 | 0,95 | 0,99 | 1,02 | 1,06 | 1,09 | 1,12 | 1,15 | 1,17 | 1,19 | 1,21 | 1,23 | |
| 18° | 0,42 | 0,45 | 0,47 | 0,50 | 0,53 | 0,56 | 0,60 | 0,63 | 0,65 | 0,68 | 0,70 | 0,72 | 0,75 | 0,76 | 0,77 | 0,79 | 0,81 | 0,82 | |
| 19º | 0,21 | 0,23 | 0,24 | 0,26 | 0,28 | 0,29 | 0,30 | 0,31 | 0,32 | 0,33 | 0,34 | 0,35 | 0,36 | 0,37 | 0,38 | 0,40 | 0,41 | 0,42 | |
| 20° | |||||||||||||||||||
| 21° | Restar | 0,21 | 0,23 | 0,24 | 0,26 | 0,28 | 0,30 | 0,31 | 0,32 | 0,33 | 0,34 | 0,35 | 0,36 | 0,37 | 0,38 | 0,38 | 0,39 | 0,39 | 0,40 |
| 22° | 0,44 | 0,46 | 0,48 | 0,51 | 0,54 | 0,57 | 0,60 | 0,62 | 0,65 | 0,67 | 0,69 | 0,71 | 0,73 | 0,74 | 0,76 | 0,78 | 0,79 | 0,81 | |
| 23° | 0,67 | 0,71 | 0,74 | 0,78 | 0,82 | 0,86 | 0,90 | 0,94 | 0,98 | 1,00 | 1,03 | 1,06 | 1,08 | 1,11 | 1,14 | 1,16 | 1,19 | 1,21 | |
| 24° | 0,89 | 0,94 | 0,99 | 1,04 | 1,09 | 1,14 | 1,21 | 1,27 | 1,31 | 1,35 | 1,38 | 1,42 | 1,46 | 1,50 | 1,53 | 1,56 | 1,58 | 1,60 | |
| 25º | 1,12 | 1,18 | 1,24 | 1,32 | 1,38 | 1,44 | 1,51 | 1,60 | 1,65 | 1,69 | 1,72 | 1,76 | 1,81 | 1,86 | 1,90 | 1,94 | 1,97 | 2,00 | |
| 28º | 1,36 | 1,43 | 1,51 | 1,57 | 1,65 | 1,73 | 1,82 | 1,90 | 1,97 | 2,02 | 2,06 | 2,12 | 2,18 | 2,23 | 2,28 | 2,31 | 2,35 | 2,39 | |
| 27° | 1,60 | 1,68 | 1,76 | 1,85 | 1,93 | 2,02 | 2,12 | 2,22 | 2,30 | 2,36 | 2,42 | 2,47 | 2,53 | 2,59 | 2,64 | 2,70 | 2,74 | 2,78 | |
| 28° | 1,83 | 1,92 | 2,02 | 2,11 | 2,21 | 2,31 | 2,43 | 2,54 | 2,63 | 2,70 | 2,76 | 2,83 | 2,96 | 2,96 | 3,02 | 3,07 | 3,12 | 3,16 | |
| 29° | 2,08 | 2,17 | 2,28 | 2,39 | 2,50 | 2,62 | 2,73 | 2,85 | 2,96 | 3,05 | 3,11 | 3,18 | 3,25 | 3,32 | 3,39 | 3,45 | 3,51 | 3,56 | |
| 30° | 2,33 | 2,45 | 2,55 | 2,67 | 2,79 | 2,91 | 3,04 | 3,16 | 3,28 | 3,38 | 3,46 | 3,54 | 3,61 | 3,69 | 3,76 | 3,83 | 3,89 | 3,95 | |
6. EXTRACTO SECO TOTAL
6.1 Principio.
El extracto seco total o materias secas totales es el conjunto de todas las sustancias que, en condiciones físicas determinadas, no se volatilizan. Estas condiciones físicas deben fijarse de tal manera que las sustancias componentes de este extracto sufran el mínimo de alteración.
El extracto seco total se calcula indirectamente conociendo la densidad del «residuo sin alcohol», que es el vino cuyo alcohol ha sido evaporado y después se ha restablecido el volumen primitivo por adición de agua.
6.2 Procedimiento.
Llevar el residuo de la destilación del alcohol del vino (sin previa neutralización) al matraz en donde se midió el vino. Lavar el matraz en donde se destiló el vino con varias porciones de agua, y llenar con estas aguas hasta volumen, el matraz con el residuo de la destilación, a la misma temperatura a la que se enrasó el vino. Mezclar bien, verter en una probeta y determinar la densidad como en 4(a).
6.3 Cálculo.
Calcular el extracto seco expresado en g/l a partir de la densidad del residuo sin alcohol utilizando la tabla 8.1.
6.3.1 Determinar la densidad del residuo sin alcohol como en 6.2.
6.3.2 Calcular la densidad 20/20 del residuo sin alcohol según la fórmula de Tabarié:
df = dv ‒ da + 1
df = densidad 20/20 del residuo sin alcohol.
dv = densidad del vino a 20º.
da = densidad a 20º de la mezcla hidroalcohólica del mismo grado que el vino.
6.3.3 Calcular la densidad del residuo sin alcohol a partir de las masas volúmicas.
df = 1,0018 (ρv ‒ρa) + 1
ρv = masa volúmica del vino a 20`.
ρa = masa volúmica a 20` de la mezcla hidroalcohólica del mismo grado que el vino.
Para valores de ρv menores a 1,05 puede tomarse 1 en vez de 1,0018.
6.4 Observaciones.
El extracto seco se expresa por la cantidad de sacarosa que, disuelta en agua hasta 1 l, da una solución de la misma densidad que el residuo sin alcohol. En la tabla 6.1 se encuentra la equivalencia de esta densidad, en extracto, expresada en sacarosa.
6.5 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A3, 1-8. 1969.
TABLA 6.1
Contenido en extracto seco (g/l)
| Densidad con dos decimales | Ferrer decimal de la densidad | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||||||||||
| Gramos de extracto por litro | |||||||||||||||||||
| 1,00 | 0 | 2,6 | 5,1 | 7,7 | 10,3 | 12,9 | 15,4 | 18,0 | 20,6 | 23,2 | |||||||||
| 1,01 | 25,8 | 28,4 | 31,0 | 33,6 | 36,2 | 38,8 | 41,3 | 43,9 | 46,5 | 49,1 | |||||||||
| 1,02 | 51,7 | 54,3 | 56,9 | 59,5 | 62,1 | 64,7 | 67,3 | 69,9 | 72,5 | 75,1 | |||||||||
| 1,03 | 77,7 | 80,3 | 32,9 | 85,5 | 88,1 | 90,7 | 93,3 | 95,9 | 98,5 | 101,1 | |||||||||
| 1,04 | 103,7 | 106,3 | 109,0 | 111,6 | 114,2 | 116,8 | 119,4 | 122,0 | 124,6 | 127,2 | |||||||||
| 1,05 | 129,8 | 132,4 | 135,0 | 137,6 | 140,3 | 142,9 | 145,5 | 148,1 | 150,7 | 153,3 | |||||||||
| 1,06 | 155,9 | 158,6 | 161,2 | 163,8 | 166,4 | 169,0 | 171,6 | 174,3 | 176,9 | 179,5 | |||||||||
| 1,07 | 182,1 | 184,8 | 187,4 | 190,0 | 192,6 | 195,2 | 197,8 | 200,5 | 203,1 | 205,8 | |||||||||
| 1,08 | 208,4 | 211,0 | 213,6 | 216,2 | 218,9 | 221,5 | 224,1 | 226,8 | 229,4 | 232,0 | |||||||||
| 1,09 | 234,7 | 237,3 | 239,9 | 242,5 | 245,2 | 247,8 | 250,4 | 253,1 | 255,7 | 258,4 | |||||||||
| 1,10 | 261,0 | 263,6 | 266,3 | 268,9 | 271,5 | 274,2 | 276,8 | 279,5 | 282,1 | 284,8 | |||||||||
| 1,11 | 287,4 | 290,0 | 292,7 | 295,3 | 298,0 | 300,6 | 303,3 | 305,9 | 308,8 | 311,2 | |||||||||
| 1,12 | 313,9 | 316,5 | 319,2 | 321,8 | 324,5 | 327,1 | 329,8 | 332,4 | 335,1 | 337,8 | |||||||||
| 1,13 | 340,4 | 343,0 | 345,7 | 348,3 | 351,0 | 353,7 | 356,3 | 359,0 | 361,6 | 364,3 | |||||||||
| 1,14 | 366,9 | 369,6 | 372,3 | 375,0 | 377,6 | 380,3 | 382,9 | 385,6 | 388,3 | 390,9 | |||||||||
| 1,15 | 393,6 | 396,2 | 398,9 | 401,6 | 404,3 | 406,9 | 409,6 | 412,3 | 415,0 | 417,6 | |||||||||
| 1,16 | 420,3 | 423,0 | 425,7 | 428,3 | 431,0 | 433,7 | 436,4 | 439,0 | 441,7 | 444,4 | |||||||||
| 1,17 | 447,1 | 449,8 | 452,4 | 455,2 | 457,8 | 480,5 | 463,2 | 465,9 | 468,6 | 471,3 | |||||||||
| 1,18 | 473,9 | 476,6 | 479,3 | 482,0 | 484,7 | 487,4 | 490,1 | 492,8 | 495,5 | 498,2 | |||||||||
| 1,19 | 500,9 | 503,5 | 506,2 | 508,9 | 511,6 | 514,3 | 517,0 | 519,7 | 522,4 | 525,1 | |||||||||
| 1,20 | 527,8 | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | ‒ | |||||||||
TABLA INTERCALAR
| 4.º decimal de la densidad | Extracto g/l |
|---|---|
| 1 | 0,3 |
| 2 | 0,5 |
| 3 | 0,8 |
| 4 | 1,0 |
| 5 | 1,3 |
| 6 | 1,6 |
| 7 | 1,8 |
| 8 | 2,1 |
| 9 | 2,3 |
7(a). AZÚCARES REDUCTORES
(Defecación plúmbica) (Provisional)
7(a).1 Principio.
Eliminación previa de todas las materias reductoras distintas de los azúcares por defecación con acetato neutro de plomo, y valoración basada en la acción reductora sobre solución cuproalcalina. Aplicable a vinos blancos y tintos de color no muy intenso.
7(a).2 Material yaparatos.
7(a).2.1 Erlenmeyer de 300 ml con refrigerante de reflujo.
7(a).2.2 Filtro de vidrio G, con capa fina de amianto de fibra fina y lavado con agua hirviendo.
7(a).2.3 Kitasatos de 500 ml con frasco de seguridad intermedio.
7(a).3 Reactivos.
7(a).3.1 Solución de acetato neutro de plomo (aproximadamente saturada). Añadir a 250 g de acetato neutro de plomo agua caliente hasta 0,5 l y agitar hasta disolución completa.
7(a).3.2 Solución de NaOH, N.
7(a).3.3 Carbonato de calcio.
7(a).3.4 Solución cúprica A. Mezclar 40 g de sulfato de cobre puro CuSO4 ∙ 5H2O con 2 ml de ácido sulfúrico puro y añadir agua hasta 1 l.
7(a).3.5 Solución tártrico-alcalina B. Mezclar 200 g de tártrato sódico-potásico (sal de Seignette) KNaC4H4O6 ∙ 4H2O con 150 g de hidróxido sódico puro y añadir agua hasta 1 l.
7(a).3.6. Solución férrica C. Mezclar 50 g de sulfato férrico puro con 20 g de ácido sulfúrico puro y añadir agua hasta 1.000 mililitros. Comprobar esta solución poniendo en un vaso de vidrio 30 ml de solución C, 100 ml de agua destilada y una gota de ortofenantrolina ferrosa. La coloración amarillo-verdosa de esta mezcla debe virar a verde puro con una sola gota de permanganato 0,1 N. En caso contrario, añadir permanganato 0,1 N a la solución hasta conseguir el viraje descrito. Generalmente es necesario añadir 1-3 ml de MnO4K 0,10 N por l de solución C.
7(a).3.7 Solución de ortofenantrolina ferrosa. Mezclar 0,695 g de sulfato ferroso, 100 ml de agua y 1,485 g de ortofenantrolina.
7(a).3.8 Solución 0,1 N de permanganato potásico. Disolver 3,16 g de permanganato potásico puro en agua hasta 1 l. Comprobar esta solución por valoración con una solución de oxalato sódico.
7(a).4 Procedimiento.
7(a).4.1 Vinos secos.‒Llevar 100 ml de vino a un matraz aforado de 125 ml, añadir (V-0,5) ml de solución de NaOH N, siendo V el volumen de solución N/10 utilizada para valorar la acidez total de 10 ml de vino, como en 20.4. Añadir agitando 5 ml de solución saturada de acetato de plomo y 1 g de carbonato de calcio, agitar varias veces y dejar sedimentar por lo menos 15 minutos, enrasar con agua destilada, añadir 0,6 ml más de agua y filtrar.
7(a).4.2 Vinos dulces.‒En un matraz aforado de 100 ml colocar un volumen de vino variable según su riqueza en azúcar. Para vinos dulces con masa volúmica comprendida entre 1,005 y 1,038, tomar 20 ml de líquido a analizar diluido al 20 por 100. Para vinos abocados con masa volúmica entre 0,997 y 1,006, tomar 20 ml de vino no diluido.
Añadir 0,5 g de carbonato de cal, unos 60 ml de agua y cantidad (0,5-2,0 ml) de solución saturada de acetato de plomo hasta el límite del enturbiamiento, agitar y dejar reposar durante 15 minutos por lo menos, agitando de cuando en cuando. Enrasar, añadir 0,2 ml más de agua y filtrar.
Un ml del filtrado corresponde a 0,04 ml de vino dulce y 0,20 mililitros de vino abocado.
En el erlenmeyer de 300 ml, poner 20 ml de solución cúprica A, 20 ml de solución tártrato-alcalina B, 20 ml de solución azucarada conteniendo de 16 a 100 mg de azúcar reductor. Hacer hervir con reflujo (o con la boca del matraz tapada con un pequeño embudo) durante 3 minutos, enfriar inmediatamente bajo una corriente de agua hasta refrigeración completa, dejar posar el óxido cuproso (inclinando el erlenmeyer en un cristalizador con algodón y descansando el cuello en el borde), filtrar después por el filtro G., con la capa de amianto, ayudando la filtración con la trompa de vacío. Tener la precaución de que, en lo posible, haya una capa de líquido sobre el precipitado adherido al fondo, tanto en el filtro como en el erlenmeyer, para evitar que se oxide. Lavar el precipitado del erlenmeyer con agua destilada hirviendo y verter en el filtro, repitiendo tres veces la operación, agitar cada vez con precaución de operar rápidamente para no dejar al descubierto el precipitado de óxido cuproso. Cambiar de Kitasato y proceder a la disolución del óxido cuproso, añadiendo primero al erlenmeyer unos 10 ml de solución C de sulfato férrico, que disuelve rápidamente el precipitado y verter la solución en el filtro. Disolver el precipitado que queda en el filtro, agitando con varilla de vidrio para facilitar la disolución. Repetir tres veces estas adiciones y lavar erlenmeyer y filtro cinco veces con unos 20 ml de agua fría cada vez.
Valorar el sulfato ferroso formado, que se encuentra en el líquido del Kitasato, con solución 0,1 N de permanganato potásico, añadiendo previamente una gota de solución de ortofenantrolina ferrosa. Dar por terminada la valoración cuando el color pase de verde-naranja a verde franco.
7(a).5 Cálculo.
La cantidad de azúcar contenida en la solución azucarada utilizada se obtiene en la tabla 7(a),I.
7(a).6 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A4, 8-10, 1969.
2. Jaulmes, P. «Analyse des vins», págs. 170-171. 1951.
TABLA 7(a).I
|
KMnO4 N/10 ‒ ml |
Azúcar invertida ‒ mg |
KMnO4 N/10 ‒ ml |
Azúcar invertida ‒ mg |
|---|---|---|---|
| 4,0 | 12,4 | 16,0 | 53,5 |
| 4,2 | 13,0 | 16,2 | 54,2 |
| 4,4 | 13,0 | 16,4 | 55,0 |
| 4,6 | 14,3 | 16,6 | 55,7 |
| 4,8 | 14,9 | 16,8 | 56,4 |
| 5,0 | 15,5 | 17,0 | 57,2 |
| 5,2 | 16,2 | 17,2 | 57,9 |
| 5,4 | 16,8 | 17,4 | 58,7 |
| 5,6 | 17,5 | 17,6 | 59,4 |
| 5,8 | 18,1 | 17,8 | 60,1 |
| 6,0 | 18,8 | 18,0 | 61,0 |
| 6,2 | 19,4 | 18,2 | 61,6 |
| 6,4 | 20,1 | 18,4 | 62,4 |
| 6,6 | 20,7 | 18,6 | 63,2 |
| 6,8 | 21,4 | 18,8 | 64,0 |
| 7,0 | 22,0 | 19,0 | 64,8 |
| 7,2 | 22,7 | 19,2 | 65,4 |
| 7,4 | 23,4 | 19,4 | 66,2 |
| 7,6 | 24,1 | 19,6 | 67,1 |
| 7,8 | 24,7 | 19,8 | 67,8 |
| 8,0 | 25,5 | 20,0 | 68,7 |
| 8,2 | 26,1 | 20,2 | 69,3 |
| 8,4 | 26,8 | 20,4 | 70,1 |
| 8,6 | 27,5 | 20,6 | 70,9 |
| 8,8 | 28,1 | 20,8 | 71,6 |
| 9,0 | 28,8 | 21,0 | 72,4 |
| 9,2 | 29,5 | 21,2 | 73,2 |
| 9,4 | 30,1 | 21,4 | 74,1 |
| 9,6 | 30,8 | 21,6 | 74,9 |
| 9,8 | 31,5 | 21,8 | 75,6 |
| 10,0 | 32,2 | 22,0 | 76,4 |
| 10,2 | 32,9 | 22,2 | 77,2 |
| 10,4 | 33,6 | 22,4 | 78,0 |
| 10,6 | 34,3 | 22,6 | 78,7 |
| 10,8 | 35,0 | 22,8 | 79,5 |
| 11,0 | 35,6 | 23,0 | 80,3 |
| 11,2 | 36,4 | 23,2 | 81,1 |
| 11,4 | 37,0 | 23,4 | 81,9 |
| 11,6 | 37,7 | 23,6 | 82,7 |
| 11,8 | 38,4 | 23,8 | 83,5 |
| 12,0 | 39,1 | 24,0 | 84,4 |
| 12,2 | 39,7 | 24,2 | 85,2 |
| 12,4 | 40,5 | 24,4 | 86,0 |
| 12,6 | 41,2 | 24,6 | 86,7 |
| 12,8 | 42,0 | 24,8 | 87,5 |
| 13,0 | 42,6 | 25,0 | 88,4 |
| 13,2 | 43,3 | 25,2 | 89,2 |
| 13,4 | 44,1 | 25,4 | 90,0 |
| 13,6 | 44,7 | 25,6 | 90,9 |
| 13,8 | 45,5 | 25,8 | 91,6 |
| 14,0 | 46,3 | 26,0 | 92,5 |
| 14,2 | 47,0 | 26,2 | 93,3 |
| 14,4 | 47,6 | 26,4 | 94,1 |
| 14,6 | 48,4 | 26,6 | 95,0 |
| 14,8 | 49,1 | 26,8 | 95,8 |
| 15,0 | 49,8 | 27,0 | 96,6 |
| 15,2 | 50,5 | 27,2 | 97,3 |
| 15,4 | 51,3 | 27,4 | 98,2 |
| 15,6 | 52,1 | 27,6 | 99,1 |
| 15,8 | 52,7 | 27,8 | 99,9 |
7(b). AZÚCARES REDUCTORES
(Defecación mercúrica) (Provisional)
7(b).1 Principio.
Eliminación previa de todas las materias reductoras distintas de los azúcares por defecación con acetato de mercurio, y valoración por la acción reductora de los azúcares sobre la solución cupro-alcalina. Aplicable a vinos jóvenes, de color intenso y ricos en azúcar, o cuando se va a realizar una valoración polarimétrica. Este método proporciona la menor pérdida de azúcares reductores.
7(b).2 Material y aparatos.
7(b).2.1 Aparato generador de ácido sulfhídrico. Filtrar el gas producido por la acción del ácido clorhídrico diluido sobre el sulfuro de hierro por paso a través de lana de vidrio y lavado en un frasco lavador con agua.
7(b).2.2 Dispositivo para eliminar el exceso de ácido sulfhídrico. Eliminar el ácido sulfhídrico en exceso después de la precipitación del exceso de mercurio, mediante aspiración conectada al tubo lateral de un Kitasato, en el que se encuentra el líquido a tratar. Tapar el matraz con un tapón atravesado por un tubo que se sumerge en el líquido. Humedecer el aire aspirado que entra en el tubo por barboteo en un matraz de un litro lleno hasta la mitad de agua.
7(b).3 Reactivos.
7(b).3.1 Solución mercúrica. Mezclar 240 g de óxido de mercurio con 240 ml de ácido acético puro y añadir agua hasta 1 l. Hervir hasta disolución completa, enfriar y añadir agua hasta un litro.
7(b).3.2 Solución de NaOH, aproximadamente 4 N. Esta solución debe poder neutralizar exactamente un volumen igual al suyo, de la solución de mercurio en presencia de fenolftaleína. Se debe comprobar esta correspondencia antes de su empleo, en particular si se observa un depósito en la solución mercúrica y si es necesario diluir la solución sódica.
7(b).3.3 Polvo de Zn. Para eliminar el exceso de mercurio.
7(b).3 (4, 5, 6, 7 y 8). Como 7(a).3 (4, 5, 6, 7 y 8).
7(b).4 Procedimiento.
7(b).4.1 Vinos secos y dulces, en los que se propone determinar el azúcar y la desviación polarimétrica. Colocar 100 ml de vino en una cápsula de 10-12 cm de diámetro y adicionar (V-0,5) ml de NaOH N, siendo V el volumen de NaOH N/10 empleado para valorar la acidez total de 10 ml de vino, como en 20.4. Evaporar en bario de agua hirviendo con corriente de aire caliente sobre la superficie del líquido, hasta reducir el volumen a 50 ml, aproximadamente. Llevar a un matraz de 100 ml, lavar tres veces la cápsula con agua, añadir 5 ml de solución mercúrica y, agitando, 5 ml de solución de hidróxido de sodio que se corresponde, volumen a volumen, con la mercúrica (solución de NaOH aproximadamente 4 N), dejar reaccionar 5 minutos por lo menos; añadir 2 ml de solución de acetato de mercurio y enrasar a 100 ml. Filtrar y recoger el filtrado en un Kitasato de 500 ml.
Tratar el filtrado con una corriente de ácido sulfhídrico para precipitar el mercurio en forma de sulfuro negro (este tratamiento puede detenerse cuando el precipitado tome color negro). Después se hace barbotar aire (previamente saturado de vapor de agua) a través del líquido, hasta que haya perdido todo su olor a sulfhídrico (5-10 minutos) y se filtra. Un ml del filtrado corresponde a 1 ml de vino. Este líquido se emplea sin dilución cuando se trata de vino seco o con unos 5 g/l de azúcar.
Con vino más rico en azúcar conviene diluir después del examen polarimétrico y antes del análisis químico.
7(b).4.2 Vinos secos y dulces en los que solamente se va a realizar el análisis del azúcar por el método químico y no por polarografía. Tomar 20 ml de vino sin diluir o diluido según norma anterior, añadir 20 ml de agua y 1 ml de solución de acetato de mercurio y añadir, agitando, 1 ml de la solución sódica.
Dejar actuar 5 minutos por lo menos, añadir 1 ml de solución mercúrica, enrasar con agua y filtrar. Tratar con ácido sulfhídrico y aire húmedo barbotando en la forma antes expuesta, y filtrar. Continuar como en 7(a).4 después de la defecación.
7(b).5 Observaciones.
En los análisis de azúcar por el método químico, para vinos no muy ricos en azúcar, en vez de eliminar el exceso de mercurio por el ácido sulfhídrico, es más práctico eliminarlo mediante la adición del polvo de cinc, de la siguiente forma: Añadir 1 g de polvo de Zn al líquido filtrado, agitar durante 2 horas de cuando en cuando, y filtrar.
7(b).6 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A4, 6-8. 1969.
2. Jaulmes, P. «Analyse des vins», pág. 176, 1951.
8. GLICEROL
8.1 Principio.
El glicerol separado del vino, previa defecación, por extracción con acetona, se oxida con ácido periódico a formaldehido. La reacción coloreada del formaldehido con la floroglucina permite la determinación espectrofotométrica a 480 nm. La presencia de sorbitol y de manitol son causa de error en la determinación del glicerol.
8.2 Material y aparatos.
8.2.1 Aparato de destilación. Compuesto de las siguientes partes:
8.2.1.1 Matraz esférico de 500 ml.
8.2.1.2 Columna rectificadora.
8.2.1.3 Columna Widmer con camisa de aislamiento.
8.2.1.4 Termómetro con escala de 0-110 °C.
8.2.1.5 Refrigerante.
8.2.2 Frascos con tapón esmerilado de 1 l y de 60 ml.
8.2.3 Matraz aforado de 50 ml.
8.2.4 Matraz aforado de 200 ml.
8.2.5 Matraces con tapón esmerilado de 60 ml.
8.2.6 Matraces aforados de 100 ml.
8.2.7 Kitasato o recipiente para vacío de 250 ml.
8.2.8 Filtro de porosidad número 4.
8.2.9 Filtros de papel Schleicher et Schull 1574 de 12,5 cm de diámetro o similar.
8.2.10 Espectrofotómetro con cubetas de 1 cm de espesor interior.
8.3 Reactivos.
8.3.1 Hidróxido de bario puro no pulverizado.
8.3.2 Arena lavada.
8.3.3 Acetona destilada.
8.3.4 Solución 1 N de hidróxido de sodio.
8.3.5 Piedra pómez.
8.3.6 Solución 1 N de ácido sulfúrico.
8.3.7 Solución 0,05 M de ácido periódico. En un matraz aforado de 1 l, introducir 11,5 g de periodato de potasio KIO4 y aproximadamente unos 800 a 850 ml de agua destilada. Si la sal utilizada es K2H3IO6 se pesan 15,2 g. Añadir en seguida 15 ml de ácido sulfúrico puro y calentar muy ligeramente en baño de agua hasta disolución completa. Enfriar a la temperatura ordinaria y llenar hasta volumen.
8.3.8 Solución de floroglucina 0,2 por 100. Esta solución debe ser renovada cada 15 días.
8.3.9 Solución madre de glicerol. Diluir unos 20 ml de glicerol bidestilado a 100 ml. Determinar después la concentración exacta de esta solución midiendo su índice de refracción, y después, por diluciones convenientes y sucesivas, llevar su concentración a 0,1 g por 1.
8.4 Procedimiento.
Extraer los polioles y valorar el glicerol.
8.4.1 Separación de polioles.‒Introducir en un matraz de 100 ml, de tapón esmerilado, 5 g de hidróxido de bario no pulverizado, 5 g de arena lavada y 10 ml de vino, que se deja deslizar por la pared del matraz.
Si el vino contiene más de 120 g de azúcar por litro, se opera sólo con 5 ml.
Agitar enérgicamente durante 30 segundos y dejar en reposo, agitando de cuando en cuando. Añadir después 50 ml de acetona y agitar otra vez durante 30 segundos. Llevar el matraz a un termostato a 45 °C durante 5 minutos, agitando enérgicamente cada minuto.
Filtrar la solución caliente por un crisol filtrante de porosidad número 4 dispuesto sobre Kitasato de vidrio en el que se han puesto 40 ml de agua y 5 ml de solución 1 N de hidróxido de sodio. Lavar el residuo tres veces con acetona, removiendo el precipitado con una espátula. Escurrir después el precipitado lo más posible. Trasvasar después este precipitado al matraz del aparato destilador, enjuagando bien el Kitasato con acetona.
Destilar la acetona con llama fuerte, hasta que el termómetro marque 100 °C, y destilar después 20 ml más de agua.
Añadir el residuo de la destilación aún caliente, 5 ml de ácido sulfúrico 1 N, dejar enfriar y trasvasar a matraz aforado de 50 ml, Se enrasa con las aguas de lavado del matraz, agitar y filtrar por filtro de papel.
8.4.2 Valoración del glicerol.‒Esta operación hay que realizarla antes de las 2 ó 3 horas siguientes a la separación de los polioles.
Llevar una alícuota de 5 ml del filtrado anterior a matraz aforado de 100 ml y enrasar con agua destilada.
Introducir 20 ml de esta dilución en un matraz de 60 ml con tapón esmerilado. Añadir 10 ml de solución 0,05 M de ácido periódico. Agitar y dejar en reposo 5 minutos.
Añadir después sucesiva y rápidamente, deslizando a lo largo de la pared, las siguientes soluciones: 10 ml de solución N de hidróxido de sodio y 10 ml de solución de floroglucina. Agitar después de la adición de cada una de las soluciones.
Llevar rápidamente la mezcla de soluciones a la cubeta del espectrofotómetro y medir la absorbancia a 480 nm en un minuto, porque la coloración violeta que se obtiene es muy fugaz.
Ajustar el espectrofotómetro de forma que la solución obtenida, al sustituir en operaciones anteriores los 5 ml de filtrado por 5 ml de agua destilada, dé una densidad óptica 0.
8.4.3 Obtención de la curva patrón.‒Diluir 25, 50, 75, 100, 125, 150, 175 ml de solución inicial de glicerol de 0,1 g por l, a 200 mililitros.
Tomar después 20 ml de cada una de estas diluciones y 20 ml de la solución inicial, y llevar a frascos de 60 ml con tapón esmerilado y tratar como en 8.4.2 para los 20 ml de la dilución del filtrado. Medir después las densidades ópticas correspondientes, ajustando el espectrofotómetro como en 8.4.2.
Esta escala corresponde a riquezas en glicerol en 0,2 ml de vino, que varían de 0 a 2 mg.
8.5 Cálculo.
Calcular el contenido en glicerol expresado en g/l.

P = peso en g de glicerol obtenido de la curva patrón.
V = volumen en ml de la muestra de vino.
f = l para contenido de azúcar en vinos inferiores a 5 g/l.
f = valor del factor de corrección según la tabla 8.1 para contenidos de azúcar en vinos superiores a 5 g/l.
8.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A21, 1-9, 1969.
TABLA 8.1
FACTORES DE CORRECCIÓN PARA EL CONTENIDO EN GLICEROL DE VINOS QUE CONTENGAN AZÚCARES
|
Contenido en azúcar ‒ g/l |
Factor |
Contenido en azúcar ‒ g/I |
Factor |
|---|---|---|---|
| 5 | 1,020 | 80 | 1,123 |
| 10 | 1,030 | 90 | 1,134 |
| 20 | 1,045 | 100 | 1,145 |
| 30 | 1,060 | 110 | 1,156 |
| 40 | 1,073 | 120 | 1,168 |
| 50 | 1,086 | 130 | 1,180 |
| 60 | 1,099 | 140 | 1,191 |
| 70 | 1,111 | 150 | 1,202 |
9. 2-3 BUTANODIOL
9.1 Principio.
El 2-3 butanodiol separado del vino, previa defecación, por extracción con acetona, se oxida con ácido periódico a acetaldehido. La reacción coloreada del acetaldehido con el nitroprusiato de sodio y la piperidina permite la determinación espectrofotométrica a 570 nm.
9.2 Material y aparatos.
9.2 (1, 2, 3, 4, 5, 6, 7, 8, 9 y 10), como 8.2. (1, 2, 3, 4, 5, 6, 7, 8, 9 y 10).
9.3 Reactivos.
9.3 (1, 2, 3, 4, 5, 6 y 7), como 8.3. (1, 2, 3, 4, 5, 6 y 7).
9.3.8. Solución de acetato sódico al 27 por 100.
9.3.9 Solución de nitroprusiato de sodio al 2 por 100. Conservar en la oscuridad y renovar semanalmente.
9.3.10 Solución de piperidina al 10 por 100. Renovar cada 15 días.
9.3.11 Solución madre de 2-3 butanodiol. Utilizar 2-3 butanodiol rectificado, desechando el primero y el último tercio. Pesar con exactitud 2 g y disolverlos en 1.000 ml. Diluir esta solución hasta 1/10.
9.4 Procedimiento.
Extraer los polioles y valorar el 2-3 butanodiol.
9.4.1 Separación de polioles.‒Como en 8.4.1.
9.4.2 Valoración del 2-3 butanodiol.‒Esta operación hay que realizarla antes de las 2 ó 3 horas siguientes a la separación de los polioles.
Llevar a un matraz de 60 ml con tapón esmerilado 5 ml del filtrado, 5 ml de solución de acetato de sodio al 27 por 100 y 10 ml de solución 0,05 M de ácido periódico.
Agitar y dejar 2 minutos en contacto. Añadir deslizando por las paredes del frasco: 5 ml de solución de nitroprusiato de sodio al 2 por 100 y 5 ml de la solución de piperidina al 10 por 100 Agitar después de la adición de cada una de las soluciones. Llevar inmediatamente la solución a la cubeta de 1 centímetro y hacer la lectura en el espectrofotómetro a 570 nm. La coloración violeta que se obtiene con la reacción alcanza el máximo de intensidad a los 30-40 segundos y después disminuye rápidamente.
Ajustar el espectrofotómetro de forma que la solución obtenida al sustituir en las operaciones anteriores los 5 ml de filtrado por 5 ml de agua destilada de una densidad óptica 0.
9.4.3 Obtención de la curva patrón.‒Diluir 10, 20, 30, 40, 50, 60 y 70 ml de la solución inicial hasta 100 ml. Tomar 5 ml de cada una de estas diluciones y tratarlas como en 9.4.2 para los 5 ml del filtrado inicial. Determinar inmediatamente las densidades ópticas máximas correspondientes, ajustando el espectro-fotómetro como en 8.4 2.
Esta curva proporciona valores de contenido en 2-3 butanodiol en 5 ml de vino que varían de O a 0,7 mg.
9.5 Cálculo.
9.5.1 Vinos con contenidos en azúcar inferior a 5 g/l.‒La cantidad de 2-3 butanodiol en g/l de vino es igual a la cantidad de 2-3 butanodiol en mg contenido en 5 ml del filtrado.
9.5.2 Vinos cuyo contenido en azúcar está comprendido entre 5 y 120 g/l.
La cantidad calculada según 9.5.1 se corrige sumando o restando el factor encontrado en la tabla 9.1.
9.5.3 Vinos con contenidos en azúcar superiores a 120 g/1.‒(La defecación se realiza sobre 5 ml de vino). La cantidad calculada según 9.5.1 se corrige como en 9.5.2, utilizando sólo la mitad del contenido en azúcar. El resultado multiplicado por 2 es el contenido en 2-3 butanodiol en g/l de vino.
9.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A21, 1-9. 1969.
|
Butanodiol g/l |
Contenido en azúcar, g/l | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | 110 | 120 | 130 | 140 | 150 | |
| 0-0,2 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 | ‒0,035 |
| 0,3 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 | ‒0,03 |
| 0,4 | –0,03 | –0,03 | –0,03 | ‒0,03 | –0,03 | –0,03 | –0,03 | –0,03 | –0,03 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 |
| 0,5 | –0,03 | –0,03 | –0,03 | –0,03 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,01 | –0,01 | –0,02 | –0,01 |
| 0,6 | –0,03 | –0,03 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,02 | –0,01 | –0,01 | –0,01 | 0,00 | 0,00 | 0,00 | 0,00 |
| 0,7 | –0,03 | –0,02 | –0,02 | –0,02 | –0,01 | –0,01 | –0,01 | –0,01 | –0,01 | –0,01 | 0,00 | 0,00 | 0,00 | 0,00 | + 0,01 |
| 0,8 | –0,03 | –0,02 | –0,02 | –0,01 | –0,01 | –0,01 | 0,00 | 0,00 | 0,00 | 0,00 | 0,00 | + 0,01 | + 0,01 | + 0,01 | + 0,02 |
| 0,9 | –0,02 | –0,02 | –0,01 | –0,01 | 0,00 | 0,00 | + 0,01 | + 0,01 | + 0,01 | + 0,01 | + 0,01 | + 0,02 | + 0,02 | + 0,03 | + 0,03 |
| 1,0 | –0,02 | –0,01 | 0,00 | 0,00 | + 0,01 | + 0,01 | + 0,01 | + 0,02 | + 0,02 | + 0,03 | + 0,03 | + 0,03 | + 0,03 | + 0,04 | + 0,05 |
| 1,1 | –0,01 | –0,01 | 0,00 | 0,00 | + 0,01 | + 0,02 | + 0,02 | + 0,03 | + 0,04 | + 0,04 | + 0,04 | + 0,05 | + 0,05 | + 0,06 | + 0,07 |
| 1,2 | ‒0,01 | ‒0,01 | 0,00 | + 0,01 | + 0,02 | + 0,03 | + 0,03 | + 0,04 | + 0,05 | + 0,05 | + 0,05 | + 0,06 | + 0,07 | + 0,07 | + 0,08 |
| 1,3 | ‒0,01 | 0,00 | + 0,02 | + 0,02 | + 0,03 | + 0,04 | + 0,05 | + 0,05 | + 0,06 | + 0,07 | + 0,07 | + 0,08 | + 0,09 | + 0,09 | + 0,10 |
| 1,4 | 0,00 | + 0,01 | + 0,02 | + 0,03 | + 0,04 | + 0,05 | + 0,06 | + 0,07 | + 0,08 | + 0,09 | + 0,09 | + 0,10 | + 0,11 | + 0,11 | + 0,12 |
| 1,5 | 0,00 | + 0,01 | + 0,03 | + 0,04 | + 0,05 | + 0,06 | + 0,07 | + 0,08 | + 0,09 | + 0,10 | + 0,11 | + 0,12 | + 0,13 | + 0,13 | + 0,14 |
| 1,6 | + 0,01 | + 0,02 | + 0,03 | + 0,05 | + 0,06 | + 0,07 | + 0,09 | + 0,10 | + 0,11 | + 0,12 | + 0,13 | + 0,14 | + 0,15 | + 0,16 | + 0,17 |
| 1,7 | + 0,01 | + 0,03 | + 0,04 | + 0,06 | + 0,07 | + 0,09 | + 0,10 | + 0,12 | + 0,13 | + 0,14 | + 0,15 | + 0,16 | + 0,17 | + 0,18 | + 0,19 |
| 1,8 | + 0,02 | + 0,03 | + 0,05 | + 0,07 | + 0,09 | + 0,10 | + 0,12 | + 0,13 | + 0,15 | + 0,16 | + 0,18 | + 0,19 | + 0,20 | + 0,21 | + 0,22 |
| 1,9 | + 0,02 | + 0,04 | + 0,06 | + 0.08 | + 0,10 | + 0,12 | + 0,14 | + 0,15 | + 0,17 | + 0,18 | + 0,20 | + 0,21 | + 0,23 | + 0,24 | + 0,25 |
| 2,0 | + 0,02 | + 0,05 | + 0,07 | + 0,10 | + 0,12 | + 0,14 | + 0,15 | + 0,17 | + 0,19 | + 0,20 | + 0,22 | + 0,24 | + 0,26 | + 0,27 | + 0,28 |
10(a). SACAROSA
(Método cualitativo colorimétrico)
10(a).1 Principio.
El vino se defeca por acetato de plomo, magnesio y permanganato de potasio a pH = 8-9. Se trata el defecado a 100 °C con difenilamina en medio clorhídrico y acético. El producto de condensación obtenido en presencia de sacarosa se extrae por cloroformo que se colorea de azul.
10(a).2 Material y aparatos.
10(a).2.1 Baño de agua con temperatura controlada.
10(a).3 Reactivos.
10(a).3.1 Acetato neutro de plomo cristalizado.
10(a).3.2 Óxido de magnesio, pesado en polvo.
10(a).3.3 Solución de permanganato potásico al 2 por 100.
10(a).3.4 Cloroformo.
10(a).3.5 Reactivo de difenilamina. A 10 ml de solución de difenilamina al 10 pm 100 en alcohol etílico absoluto, añadir 20 ml de ácido acético glacial y 70 ml de ClH puro (d = 1,18-1,19).
Para comprobar la pureza de este reactivo añadir 2 ml de reactivo de difenilamina y 2 ml de agua destilada, calentar en baño de agua hirviendo durante 5 minutos. Enfriar la solución bruscamente por inmersión en agua fría y extraer con 1 ml de cloroformo, no debiendo presentar el líquido extraído coloración azul.
10(a).4 Procedimiento.
Disolver en 10 ml de agua 100 a 200 mg de óxido de magnesio y unos 200 mg de acetato neutro de plomo. Llevar al baño de agua que se mantiene a 90-95 °C durante 3 a 5 minutos. Añadir 2 ml del vino que debe estar limpio y con una riqueza en azúcar inferior al 1 por 100.
Diluir la muestra si la riqueza en azúcar es superior a la indicada como límite.
Comprobar si la dosis de acetato de plomo es suficiente y no excesiva, lo que disminuiría la sensibilidad de la reacción. Para ello añadir gota a gota, al líquido que sobrenada, una solución concentrada de acetato neutro de plomo, hasta que no se acuse enturbiamiento al caer la gota sobre el líquido. Añadir entonces solución saturada de sulfato de sodio para eliminar el exceso de plomo.
Añadir 0,5 ml de la solución de permanganato potásico y mantener la mezcla en el baño de agua a 90-95° durante 10 minutos.
Enfriar bruscamente por inmersión en agua fría, y filtrar. Puede aparecer una débil coloración amarilla, pero no influye en la reacción.
Poner en un tubo de ensayo 2 ml del líquido filtrado, añadir 2 ml del reactivo de difenilamina; mantener en baño de agua hirviendo, durante 5 minutos exactamente. Enfriar por inmersión en agua fría, añadir 1 ml de cloroformo y observar la coloración que se forma.
10(a).5 Expresión de los resultados.
Los vinos secos no adicionados con sacarosa pueden dar una coloración gris-azulada muy ligera, pero, en presencia de sacarosa, aparece una franca coloración azul.
Los vinos ricos en azúcar dan una coloración amarillo oro en ausencia de sacarosa, pero en presencia de ésta, la coloración es verde franco,
Los resultados son sensibles para riquezas de sacarosa desde 0,2 mg.
10(a).6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A-5, 1-2. 1969.
10(b). SACAROSA
(Método cuantitativo)
10(b).1 Principio.
La sacarosa se determina por la diferencia de los poderes reductores antes y después de la hidrólisis clorhídrica del líquido procedente de la defecación del vino.
10(b).2 Reactivos.
10(b).2.1 ClH puro.
10(b).3 Procedimiento.
Poner en dos matraces erlenmeyer de 200 ml cada uno el mismo volumen del líquido de defecación, según las normas indicadas para el análisis de azúcares. La defecación se realiza como en 7(b) con eliminación previa del alcohol.
Añadir a cada matraz un volumen de ácido clorhídrico puro a razón de 0,3 ml por 10 ml de líquido azucarado.
En uno de los matraces añadir inmediatamente 0,3 ml de Na (OH), 12 N, por cada 10 ml de líquido azucarado procediendo a la valoración de los azúcares reductores.
Llevar el otro matraz con el contenido sin neutralizar a baño de agua hirviendo durante 2 minutos. Añadir entonces el mismo volumen de Na (OH), 12 N que al otro matraz y valorar.
10(b).4 Cálculo.
La diferencia entre las cantidades de azúcares reductores encontrada en los resultados de estas dos valoraciones, multiplicada por 0,95 da la riqueza en sacarosa de la muestra en estudio.
Este resultado se expresa en g por l de vino, teniendo presentes las diluciones efectuadas eventualmente en el curso de la defecación y del volumen de la muestra de vino que se sometió a la defecación.
10(b).5 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O.I.V. A5, 4, 1969.
11. CENIZAS
11.1 Principio.
Se denominan cenizas de un vino, al conjunto de los productos de incineración del residuo de evaporación de un volumen conocido del vino, realizada de manera que se puedan obtener todos los cationes (excepto amonio) en forma de carbonatos y otras sales minerales anhidras.
11.2 Material y aparatos.
11.2.1 Horno eléctrico regulable.
11.2.2 Baño de agua y baño de arena.
11.2.3 Lámpara de infrarrojo.
11.2.4 Cápsula de platino o de cuarzo de 70 mm de diámetro y 25 mm de altura, fondo plano.
11.3 Procedimiento.
Colocar 20 ml de vino en una cápsula tarada en balanza que aprecie 1/10 de mg. Evaporar con precaución en baño de agua, después de evaporar hasta consistencia siruposa, continuar el calentamiento sobre baño de arena con moderación y durante una media hora. Es conveniente ayudar a la evaporación con la aplicación de rayos infrarrojos hasta carbonización. Cuando ya no se desprendan vapores, llevar la cápsula al horno eléctrico a 525 °C ± 25° y con aireación continua.
Después de 5 minutos de carbonización completa, sacar la cápsula del horno, dejar enfriar y añadir 5 ml de agua que se evaporan en baño de agua, y llevar de nuevo al horno a 525 °C.
Si la combustión de las partes carbonosas no se consigue en 15 minutos, volver a comenzar la operación de adición de agua, evaporación y recalcinación.
Cuando se trate de un vino rico en azúcares, se recomienda adicionar unas gotas de aceite puro vegetal al extracto, antes de comenzar la calcinación para impedir el desbordamiento de la masa del contenido. La duración de la primera carbonización deberá ser en este caso 15 minutos.
Después de enfriar en el desecador cápsula y cenizas, se pesan.
11.4 Cálculo.
Calcular el contenido en cenizas expresado en g/l.
Cenizas = 50 P g/l
P = peso en g de las cenizas contenidas en 20 ml de vino.
Dar los resultados con una aproximación de 0,03 g/I.
11.5 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A6, 1. 1969.
2. «Métodos Oficiales de Análisis de Vinos», pág. 106. Ministerio de Agricultura, 1956.
12. ALCALINIDAD DE CENIZAS
12.1 Principio.
Se denomina alcalinidad total de cenizas la suma de los cationes, diferentes del amonio, combinados con los ácidos orgánicos del vino.
La valoración se funda en la volumetría con ácido sulfúrico, valorando en retorno después de disolver en el mismo y en caliente, las cenizas, y empleando el naranja de metilo como indicador.
12.2 Material y aparatos.
12.2.1 Baño de agua.
12.2.2 Pipeta de 10 ml.
12.2.3 Varilla de vidrio.
12.2.4 Bureta de valoraciones.
12.3 Reactivos.
12.3.1 Ácido sulfúrico N/10.
12.3.2 Solución acuosa de naranja de metilo al 1 por 1.000.
12.3.3 Hidróxido sódico N/10.
12.4 Procedimiento.
Añadir a las cenizas ya pesadas, 10 ml de ácido sulfúrico N/10 y llevar la cápsula a un baño de agua hirviendo durante un cuarto de hora, frotando varias veces el fondo de la cápsula con una varilla de vidrio para activar la disolución de las partículas difíciles de disolver. Añadir en seguida dos gotas de una solución de naranja de metilo al 1 por 1.000 y valorar el exceso de SO4H2 con Na(OH) N/10, hasta que el indicador vire a amarillo.
12.5 Cálculo.
Calcular la alcalinidad de las cenizas expresadas en meq/l o en g/l de carbonato potásico.
Alcalinidad de as cenizas = 5 (10 ‒ V) meq/l
Alcalinidad de las cenizas = 0,345 (10 ‒ V g/l de carbonato potásico.
V = volumen en ml de NaOH N/10 utilizados.
Dar los resultados con una aproximación de 0,5 g por l en carbonato potásico.
12.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A7, 1-2, 1969.
13. FOSFATOS
13.1 Principio.
Oxidación del vino, evaporación e incineración. Precipitación del ácido fosfórico en medio nítrico al estado de fosfomolibdato de amonio. Esta sal se disuelve por acción de un exceso de sosa N/2 en presencia de formol. Valoración del exceso de sosa por ácido clorhídrico N/2 en presencia de fenolftaleína.
13.2 Material y aparatos.
13.2.1 Cápsula de sílice de 70 mm de diámetro.
13.2.2 Baño de arena.
13.2.3 Horno de mufla.
13.2.4 Erlenmeyer de 500 ml.
13.2.5 Vasos de precipitado y elementos para volumetría.
13.2.6. Agitador mecánico.
13.3 Reactivos.
13.3.1 Solución de ácido nítrico al 25 por 100 (d = 1,153). Añadir a 440 ml de ácido nítrico comercial (d = 1,33) agua hasta 1.000 ml.
13.3.2 Solución de nitrato amónico. Añadir, a 340 g de nitrato de amonio, agua hasta 1.000 ml.
13.3.3 Solución de molibdato de amonio. Añadir, a 30 g de molibdato de amonio, agua hasta 1.000 ml.
13.3.4 Solución de lavado. 50 g de nitrato de amino, 40 ml de ácido nítrico al 25 por 100 y agua destilada hasta 1 l.
13.4 Procedimiento.
Evaporar a sequedad 100 ml de vino en cápsula de cuarzo en sucesivas adiciones. Añadir después 4 ml de ácido nítrico y calentar progresivamente en el baño de arena y después en el horno de mufla.
Añadir a las cenizas 2 ml de ácido nítrico y evaporar a sequedad en el baño de agua para insolubilizar la sílice. Añadir 10 ml de ácido nítrico al 25 por 100; remover y arrastrar las cenizas a un filtro; lavar la cápsula y filtro con 5 ml del mismo ácido y después con 20-30 ml de agua destilada. Recoger el filtrado en un erlenmeyer de unos 500 ml, adicionar 25 ml de solución de nitrato de amonio de 340 g por l y agitar.
Después añadir de una vez 80 ml de solución de molibdato amónico del 3 por 100 y agitar mecánicamente durante 12 minutos a razón de 200 sacudidas por minuto. Decantar y filtrar por papel. Lavar luego el precipitado con 75 ml de líquido de lavado dos veces. Cada lavado no debe durar más de 10 minutos. Lavar 8 veces más con agua destilada.
Colocar el filtro y el precipitado en un vaso con 50 ml de Na(OH) N/2. Añadir 12,5 ml de formol neutralizado en presencia de fenolftaleína, para disolver el precipitado de fosfomolibdato. Escurrir el filtro y extender sobre la pared del vaso, por encima del líquido de la solución. Valorar el exceso de sosa con ClH N/2, hasta decoloración de la fenolftaleína, habiendo sumergido al final nuevamente el filtro.
13.5 Cálculo.
Calcular el contenido en fosfatos expresado en meq de ácido fosfórico (considerado como triácido), o en g de P2O5 por l de vino.
Fosfatos = 0,577 (50 — V) meq de ácido fosfórico.
Fosfatos = 0,01365 (50 — V) g de P205 por 1 de vino.
V = volumen en ml de solución N/2 de ácido clorhídrico.
Dar los resultados con una aproximación de 0,01 g/l.
13.6 Referencia.
1 Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A16, 1-2 1969.
14. CALCIO
(Método complexométrico)
14.1 Principio.
Valoración del calcio por complexometría sobre la solución nítrica o clorhídrica de las cenizas del vino.
14.2 Material y aparatos.
14.2 (1, 2, 3 y 4) como 11.2 (1, 2, 3 y 4).
14.2.5 Matraces erlenmeyer de 1.000 ml.
14.2.6 Matraces erlenmeyer de 100 ml.
14.3 Reactivos.
14.3.1 Ácido clorhídrico 0,2 N.
14.3.2 Solución de complexona III 0,05 M. Añadir a 18,61 g de complexona III (sal disódica bihidratada del ácido etilendiamino-tetracético) agua destilada hasta 1.000 ml y disolver.
14.3.3 Lejía de sosa al 40 por 100. Añadir a 40 g de hidróxido de sodio puro agua destilada hasta 100 ml.
14.3.4 Indicador de calcon. Mezclar 1 g de calcon (sal sádica del ácido 1(2 hidroxil-naltilazon2 nafto1-4 sulfónico) con 100 g de cloruro de sodio puro.
14.3.5 Solución de cloruro de calcio 0,05 M. Disolver 5,05 g de carbonato de cal puro en la cantidad necesaria de ácido clorhídrico diluido y completar a 1 litro. Valorar esta solución por medio de solución 0,05 M de complexona III en presencia de calcon en la forma que luego se describe.
14.4 Procedimiento.
Evaporar a sequedad en bario de agua hirviendo 50 ml de vino colocados en cápsulas preferentemente de platino. Incinerar el residuo como en 11. Disolver las cenizas en 10 ml de ClH 0,2 N, llevar a un matraz aforado de 50 ml; lavar varias veces la cápsula con agua destilada vertiéndola en el matraz. Enrasar y agitar.
Tomar 20 ml de la solución de cenizas y calentar hasta ebullición, en un erlenmeyer de unos 100 ml. Dejar enfriar y después añadir 0,5 mi de solución de Na(OH) al 40 por 100, 10 ml de solución de complexona III 0,05 M y 100 mg aproximadamente de indicador calcon.
Si el color de la mezcla es rojo-vinoso, añadir complexona en exceso hasta aparición de color azul-violeta.
Evitar una cantidad de complexona en exceso relativamente grande, que enmascara el punto de viraje.
Valorar el exceso de complexona III añadiendo solución 0,05 M de cloruro de calcio. El indicador calcon virará de azul violeta a rojo vinoso al final de la reacción.
14.5 Cálculo.
Calcular el contenido en calcio como iones Ca en meq/l, o como iones Ca expresados en g/l.
Calcio = (10 ‒ V) 5 meq/l de ion Ca.
Calcio = (10 ‒ V) 0,1 g/1 de ion Ca.
V = volumen en ml de la solución 0,05 M de cloruro de cal.
14.6 Referencias.
1. Castino, M. «Determinazione complessometrica del Calcio o del Magnesio nei vini». Rivista de Viticoltura e di Enologia di Conegliano, 9, 1960.
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A26, 2-3, 1969.
15. MAGNESIO
15.1 Principio.
Valoración del magnesio por complexometría sobre la solución nítrica o clorhídrica de las cenizas del vino.
15.2 Material y aparatos.
15.2 (1, 2, 3 y 4), como 11.2 (1, 2, 3 y 4). 15.2 (5 y 6), como en 14.2 (5 y 6).
15.3 Reactivos.
15.3.1 Solución de complexona III 0,05 M. Como en 14.3.2.
15.3.2 Solución tampón de pH 10. Disolver 50 g de cloruro de amonio en 350 g de solución de amoníaco al 25 por 100 y añadir agua destilada hasta 1.000 ml.
15.3.3 Indicador negro de eriocromo T. Mezclar 1 g de negro de eriocromo T, con 100 g de cloruro de sodio puro.
15.3.4 Solución 0,05 M de cloruro de magnesio. Disolver 2,25 g de óxido de magnesio puro (*) en la cantidad necesaria de ácido clorhídrico diluido y llevar después el volumen a 1 litro. Valorar esta solución con complexona III 0,05 M (15.3.1) en presencia de negro de eriocromo T (15.3.3) como se indica a continuación.
(*) El óxido de magnesio del comercio está generalmente carbonatado, aun puro, por lo que debe calcinarse. Tomar 5 g en cápsulas de platino; llevar al rojo durante más de 30 minutos. Dejar enfriar y pesar los 2,25 g.
15.4 Procedimiento.
Tomar 20 ml de la solución de cenizas antes preparada, llevar a ebullición en un erlenmeyer de unos 100 ml, dejar enfriar y añadir 10 ml de solución de complexona III 0,05 M,5 ml de solución tampón pH 10,50 mg aproximadamente de indicador negro de eriocromo T.
Valorar después el exceso de complexona III con la solución de cloruro de magnesio 0,05 M. El indicador vira del azul al rojo vinoso.
15.5 Cálculo.
Calcular el contenido en magnesio expresado en meq/l o en g/l de iones Mg.
Magnesio = (V ‒ V') x 5 meq/l.
Magnesio = (V ‒ V') X 0,06075 g/l de iones Mg.
V = volumen en ml de solución 0,05 M de cloruro de calcio utilizado para valorar el exceso de complexona III en la determinación del calcio (14.4).
V' = volumen en ml de solución de cloruro de magnesio 0,05 M.
15.6 Referencias.
1. Castino, M. «Determinazione complessometrica del Calcio o del Magnesio nei vini». Rivista di Viticoltura e di Enologia di Conegtiano, 9, 1960.
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A26, 2-3, 1969.
16. HIERRO
(Método colorimétrico del tiocianato)
16.1 Principio.
El hierro oxidado por el agua oxigenada se combina con el ion tiociánico en medio clorhídrico. El color rojo se compara con escala patrón o se mide por espectrofotometría. Para los vinos tintos, el tiocianato férrico se extrae con éter. Para valorar solamente el hierro trivalente, se suprime la adición del agua oxigenada.
16.2 Material y aparatos.
16.2.1 Espectrofotómetro o colorímetro para lectura a 508 nm de longitud de onda.
16.2.2 Tubos de 200 X 20 mm.
16.2.3 Bola de decantación de 50 ml.
16.3 Reactivos.
16.3.1 Ácido clorhídrico puro. Densidad: 1,18-1,19.
16.3.2 Agua oxigenada de 10 volúmenes.
16.3.3 Éter exento de peróxido. Destilar el éter (sulfúrico) antes de su empleo o bien mantener el éter en presencia de sodio o de carbón activado o con bióxido de manganeso.
16.3.4 Solución de tiocianato de potasio. Añadir a 200 g de tiocianato de potasio agua destilada hasta 1 litro, y disolver.
16.3.5 Solución de cloruro férrico de 0,1 g de hierro por litro. Disolver 0,1 g de hierro puro en 10 ml de CIH y 30 ml de agua. Añadir 1 ml de NO3H y hervir. Enfriar y añadir agua destilada hasta 1 litro.
También puede prepararse la siguiente solución de hierro. Disolver 0,863 g de alumbre de hierro, 0,863 g de amonio puro y 10 ml de ácido clorhídrico en 100 ml de agua y diluir hasta 1.000 ml.
16.3.6 Alcohol de 20°.
16.4 Procedimiento.
16.4.1 Determinación del hierro total.‒(Férrico-ferroso).
16.4.1.1 Vinos blancos o poco coloreados.‒Colocar 10 ml de vino en un tubo de 200 x 20 mm, añadir 1 ml de ClH, 5 gotas de agua oxigenada y 1 ml de solución de tiocianato.
A la aparición del color añadir 20 ml de éter, agitar para mezclar las dos fases sin emulsionar y dejar decantar 5 minutos. Determinar la absorbancia del líquido decantado en espectrofotómetro o colorímetro a 508 nm.
Ajustar el espectrofotómetro a cero, utilizando como prueba en blanco 10 ml de vino acidulados con 1 ml de ácido clorhídrico y diluidos con 1 ml de agua destilada. De esta forma se compensa la influencia del color del vino y de la posible aparición de color o del reavivado del vino, ambas coloraciones debidas a la acidez añadida al vino.
Obtener una curva, procediendo en la forma anteriormente descrita y utilizando en vez de 10 ml de vino mezclas de 5 ml de alcohol de 20° y porciones de 0,5; 1,0; 1,5; 2,0; 2,5 ml, completadas hasta 5 ml con agua destilada. Estas mezclas contendrán, respectivamente, 5, 10, 15, 20 y 25 mg/l de hierro.
16.4.1.2 Vinos tintos.‒Como en 16.4.1.1, pero utilizando sólo 0,2 ml de ClH. El exceso de acidez dificulta la extracción del tiocianato férrico por el éter.
16.4.2 Determinación del hierro trivalente.‒Como en 16.4.1, pero sin adicionar agua oxigenada.
18.5 Cálculo.
Calcular el contenido en hierro expresado en mg/l por comparación de la absorbancia con la curva patrón.
16.6 Observaciones.
En lugar del éter algunos autores (2) recomiendan el empleo del acetato de etilo, que no tiene el inconveniente de la posible presencia de peróxidos; sin embargo, en algunos vinos de intenso color de nuestro país se ha observado extracción de parte de la materia colorante con el acetato de etilo, lo que no sucede con el éter.
16.7 Referencias.
1. Ribereau-Gayon, J., y Peynaud, E. «Analyse et Controle dens Vins», pág. 194, 1958.
2. Jaulmes, P.; Brun, S.; Tour, C.; Cabanis, J. C. «Dosage direct du fer dans le vino». Société Pharmacie Montpellier, 24: 2, 1964.
17. COBRE
17.1 Principio.
El ditiocarbamato de sodio reacciona con el cobre (reacción de Delepine), dando la sal correspondiente a este metal y coloración amarilla oro, cuya intensidad se mide por colorimetría o espectrofotometría.
Para evitar interferencias debidas a la presencia de hierro y otros cationes polivalentes presentes en el vino se emplea la sal disódica del ácido etilendiaminotetraacético, que forma complejos solubles y muy estables a pH = 8. También puede utilizarse el citrato de amonio para evitar la interferencia del hierro.
17.2 Material y aparatos.
17.2.1 Espectrofotómetro o colorímetro con filtro para longitud de onda de 420 nm.
17.2.2 Bola de decantación de unos 100 ml.
17.3 Reactivos.
17.3.1 Solución acuosa al 0,1 por 100 de dietilditiocarbamato de sodio.
17.3.2 Suspensión de sal bisódica del ácido etilendiaminotetraacético al 20 por 100, o bien solución acuosa del citrato de amonio al 20 por 100.
17.3.3 Solución amoniacal al 15 por 100.
17.3.4 Alcohol metílico.
17.3.5 Tetracloruro de carbono.
17.3.6 Agua bidestilada en vidrio. Utilizarla también para la preparación de reactivos.
17.4 Procedimiento.
Poner en una bola de decantación 10 ml del vino, añadir 5 ml de una suspensión de la sal bisódica del ácido etilendiaminotetraacético (ó 5 ml de solución de citrato amónico) y llevar a pH 8 con la solución amoniacal. Añadir 1 ml de reactivo de dietilditiocarbamato de sodio y 5 ml de alcohol metílico (para evitar emulsión), y agitar durante 1 minuto.
Extraer varias veces con tetracloruro de carbono (agitando cada vez durante 1 minuto) hasta recoger 20 ml, cuidando que no pasen restos de agua, pues éstos dan enturbiamiento con el tetracloruro de carbono. Si pasa alguna gota, filtrar por papel de filtro puro, quedando las trazas de agua en el papel.
Determinar la absorbancia en el espectrofotómetro a 420 nm de longitud de onda.
Para la prueba en blanco utilizar los mismos reactivos en las mismas proporciones, sustituyendo el volumen de vino por otro igual de agua bidestilada en aparato de vidrio.
La curva que previamente será construida a partir de lecturas correspondientes a escala de diferentes riquezas de cobre es una línea recta, por obedecer a la ley de Lamber-Beer. En el intervalo en que se opera con 1 ml de reactivo pueden valorarse 10 p. p. m. de cobre; para valores superiores, se añadirán 2 ml o se toma menor volumen de muestra.
17.5 Cálculo.
El valor obtenido para la absorbancia a 420 nm se lleva a la curva-patrón y se expresa el correspondiente contenido en cobre en p. p. m.
17.6 Referencia.
1. Garoglio, P. G., y Stella, C..Methodi ufficiali di analiis per i mosti, i vini e gli aceti», pág. 75, 1958.
18. POTASIO
(Fotometría de llama)
18.1 Principio.
Se utiliza una solución de referencia con 100 mg de potasio por litro y con diversos aniones, cationes y materia orgánica en proporciones tales que den un compuesto similar a un vino diluido a 1/10 con agua.
18.2 Material y aparatos.
18.2.1 Fotómetro de llama y accesorios.
18.2.2 Pipetas de 10 ml.
18.2.3 Matraces aforados de 1.000 ml.
18.3 Reactivos.
18.3.1 Solución de referencia.‒Disolver 481,3 mg de tarirato ácido de potasio en 500 ml de agua muy caliente y mezclar con una solución de 10 ml de alcohol, 700 mg de ácido cítrico, 300 mg de azúcar, 1.000 mg de glicerol, 20 mg de fosfato monosódico, 10 mg de cloruro de calcio seco y 10 mg de cloruro de magnesio seco en 400 ml de agua. Completar con agua hasta 1.000 ml.
Añadir dos gotas de siotiocianato de alilo para conservación más segura de la solución.
18.3.2 Solución de dilución.‒Igual que la anterior, cambiando el tartrato ácido de potasio por 383 mg de ácido tártrico.
Para preparar la solución de referencia, comenzar por disolver 481,3 mg de tartrato ácido de potasio en medio litro de agua muy caliente y mezclar esta solución con los otros componentes, previamente disueltos en 400 ml de agua; enrasar después a 1 litro exactamente.
Para la conservación más segura de esta solución se añaden 2 gotas de sotiocianato de alilo.
Con vinos dulces, añadir una cantidad de azúcar igual a la décima parte de la que contenga el vino.
Añadir a la solución de referencia una cantidad de fosfato monosódico equivalente al contenido en sodio del vino cuando este contenido sea superior al equivalente a 20 mg de fosfato monosódico.
En el caso del fraude por adición de 1 g/l de salicilato de sodio, añadir 100 mg de salicilato de sodio por litro de la solución de referencia y de dilución. El salicilato de sodio tiene una influencia muy marcada en el resultado del análisis.
18.4 Procedimiento.
Regular el aparato y establecer una curva de calibración con la solución de referencia pura y con diversas diluciones de la solución de referencia diluida a 1/20, 1/10, 1/5, 1/2, con la solución de dilución.
Diluir el vino a 1/10 con agua y hacer la determinación en el fotómetro.
Si la lectura no queda comprendida entre los valores 40 y 100 de la escala del galvanómetro, diluir convenientemente el vino con solución de dilución.
18.5 Cálculo.
Calcular el contenido en potasio expresado en g/l con una aproximación de ± 0,02 g/l.
Potasio = 10 L g/l
L = valor correspondiente a la lectura de la escala del galvanómetro en la curva de calibración.
18.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. AS, 1-6, 1969.
19. SODIO
(Método del fotómetro de llama)
19.1 Principio.
Como 18.1, referido a sodio.
19.2 Material y aparatos.
Como en 18.2.
19.3 Reactivos.
19.3.1 Solución de referencia.‒Como en 18.3.1, sustituyendo los 20 mg de fosfato monosódico por 50,84 mg de cloruro de sodio.
19.3.2 Solución de dilución.‒Como en 18.3.2, suprimiendo los 50,84 mg de cloruro de sodio.
19.4 Procedimiento.
Como en 18.4. Para la curva de calibración, utilizar la solución de referencia diluida a 1/20, 1/10, 1/2, 3/4.
19.5 Cálculo.
Calcular el contenido en sodio expresado en g/l con una aproximación de ± 0,05 g/l.
19.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A25, 1-2, 1969.
20. ACIDEZ TOTAL
20.1 Principio.
La acidez total de un vino se considera como la suma de los ácidos titulables cuando se lleva el vino a pH = 7 por adición de un licor alcalino valorado. El ácido carbónico y el anhídrido sulfuroso libre y combinado no se consideran comprendidos en la acidez total.
El gas carbónico se elimina previamente del vino por agitación en frío y con vacío parcial.
20.2 Material y aparatos.
20.2.1 Potenciómetro con electrodo de vidrio.
20.2.2 Agitador magnético.
20.2.3 Puente con solución saturada de CIK para evitar la introducción del electrodo de calomelanos en el vino.
20.2.4 Kitasato de un litro.
20.2.5 Pipeta de 20 ml
20.2.6 Vaso de 100 ml de capacidad.
20.2.7 Bureta de 50 ml.
20.3 Reactivos.
20.3.1 Solución de Na(OH) N/10.
20.4 Procedimiento.
Poner 50 ml de vino en un Kitasato de 1 l de capacidad, conectar al vacío agitando al mismo tiempo el matraz. El desprendimiento de CO2 se aprecia a los pocos momentos. Observar atentamente el momento en que dejan de desprenderse las burbujas, desconectar del vacío.
Tomar 20 ml del vino sin CO2 y llevar a un vaso de unos 100 ml de capacidad, introducir el electrodo de vidrio en el vino y el de calomelanos en vaso lateral con solución saturada de ClK. Disponer un puente de la misma solución en tubo de vidrio con los extremos uno en cada vaso estableciendo contacto.
Poner en marcha el agitador y añadir desde bureta solución N/10 de sosa al vino hasta que la aguja del potenciómetro marque pH = 7, manteniendo el agitador en marcha durante la operación, que no debe durar más de cinco minutos.
20.5 Cálculo.
Calcular la acidez total expresada en meq/l con una aproximación de 0,1 meq/l o en g de ácido tártrico.


V = volumen en ml de Na (OH) N/10.
V' = volumen en ml de iodo N/100 utilizado para la oxidación de anhídrido sulfuroso libre (21.‒).
V" = volumen en ml de iodo N/100 utilizado para la oxidación del anhídrido sulfuroso combinado (21.‒).
20.6 Referencias.
1. Recueil des Méthodes Internacionales d'Analyse des Vins. O. I, V. A10, 1-3, 1969.
2. Jaulmes, P. «La mesure de l'acidite total des vins». Annal des Falsifications et des Fraudes, 556, 1955.
21. ACIDEZ VOLÁTIL
21.1 Principio.
La acidez volátil está constituida por la parte de ácidos grasos pertenecientes a la serie acética que se encuentra en los vinos, ya sea en estado libre o de sal. Se determina mediante la separación de los ácidos volátiles por arrastre con vapor de agua y rectificación de los vapores. Se debe evitar con precaución la presencia de gas carbónico en el destilado. La acidez del anhídrido sulfuroso libre y combinado arrastrados con el destilado no deben comprenderse en la acidez volátil, por lo que hay que restar la equivalencia de su acidez de la del destilado, así como la del ácido sórbico eventualmente presente. Para estas correcciones se sigue la norma Jaulmes (1), en la que se considera como completa la influencia de SO2 libre y sólo la mitad de la del combinado.
21.2 Material y aparatos.
21.2.1 Aparato para destilación aislada (fig. 21.1).
21.2.1.1 Matraz generador de vapor de 3.500 ml.
El agua condensada en el tubo de unión del matraz con el barbotador se purga por el purgador (c).
21.2.1.2 Barbotador (b).‒Tubo cilíndrico de 3 cm de diámetro y 27 cm de altura. Este tubo reposa sobre un disco de amianto de 15 cm de diámetro, con un orificio central de 29 mm. El tubo (d) por el que entra el vapor en el barbotador debe llegar a 1 cm del fondo.
21.2.1.3 Columna rectificadora (e).‒Constituida por un tubo cilíndrico de 20 mm de diámetro y 50 cm de altura, conteniendo una hélice de tela de acero inoxidable número 100 plegada, con 15 mm de paso.
21.2.1.4 Refrigerante de West (f).‒De 40 cm de longitud activa, colocado verticalmente.
21.3 Reactivos.
21.3.1 Solución de Na (OH) N/10.
21.3.2 Solución de fenolftaleína al 1 por 100 en alcohol neutro.
21.3.3 Solución de yodo N/100.
21.3.4 Solución de engrudo de almidón.
21.4 Procedimiento.
Alimentar el generador de vapor con agua de cal o de barita limpia, manteniéndolo lleno hasta unos 2/3 de su volumen. Poner en el barbotador 20 ml del vino exento de gas carbónico como en 20.4. Añadir al vino unos 0,5 g de ácido tártrico, poner en marcha el generador de vapor, manteniendo abierta la salida del tubo purgador del vapor; después de cerrar ésta, calentar el barbotador. Durante la operación se regula el calentamiento de forma que el volumen de líquido en el barbotador no pase sensiblemente de los 20 ml iniciales.
Destilar en unos 12-5 minutos 250 ml.
Valorar con solución de sosa N/10 en presencia de dos gotas de fenolftaleína como indicador.
Valorar el sulfuroso libre en este destilado, añadiendo al terminar la anterior valoración una gota de ácido clorhídrico puro, para acidular nuevamente y valorar el SO2 libre con solución de iodo N/100, añadiendo también 2 ml de solución de almidón como indicador y un cristal de ioduro de potasio.
Para determinar el ácido sulfuroso combinado con el acetaldehído, añadir 20 ml de solución saturada de bórax (el líquido toma un color rosa pálido) y valorar nuevamente con solución de iodo N/100.
21.5 Cálculo.
Calcular la acidez volátil expresada en meg/l, con una aproximación de 0,2 ml, o en g/l de ácido sulfúrico o de ácido acético, con una aproximación de 0,1 g/l.
Acidez volátil = 5 (V ‒ V'/10 ‒ V''/20) meq/l


'V = volumen en ml de Na (OH) N/10.
V' = volumen en ml de iodo N/100 utilizado en la oxidación del anhídrido sulfuroso libre.
V" = volumen en ml de iodo N/100 utilizado en la oxidación del anhídrido sulfuroso combinado con el acetaldehído.
21.6 Observaciones.
En la destilación del vino para la determinación de la acidez volátil el ácido sórbico pasa casi en su totalidad al destilado, junto con el ácido acético, falseando el resultado del análisis. Para corregir el resultado, determinar el ácido sórbico en el destilado como en 29, procediendo en la siguiente forma simplificada: tomar 0,5 ml del destilado de la acidez volátil, llevar a la cubeta de 1 cm de espesor interno, añadir 1,5 ml de la solución (a), dejar la cubeta al aire durante unos minutos y medir después la absorbancia en el espectrofotómetro a 256 mμ. Graduar el espectrofotómetro con 0,5 ml de la solución (b) y 1,5 ml de la solución (a).
21.7 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. All, 1-4, 1969.
2. Jaulmes, P. Analyse des Vins, 2.° ed., 1951.
22. ÁCIDO SÓRBICO EN EL DESTILADO PARA ACIDEZ VOLÁTIL
22.1 Principio.
En la destilación del vino para la determinación de la acidez volátil el ácido sórbico pasa casi en su totalidad al destilado junto con el ácido acético, falseando el resultado del análisis. Para valorar este ácido sórbico se parte de una muestra de 0,5 ml de destilado (lo que no supone un error sensible) y se valora por espectrofotometría en el ultravioleta.
22.2 Material y aparatos.
22.2.1 Espectrofotómetro ultravioleta.
22.2.2 Cubetas de cuarzo de 1 cm.
22.2.3 Pipetas de 0,5 ml y 1,5 ml.
22.3 Reactivos.
22.3.1 Solución a): Mezclar 0,5 g de NaHCO3 con 0,001 g de CuSO4 ∙ 5H2O y añadir agua hasta un litro.
22.3.2 Solución b): Llevar 20 mg de ácido sórbico a matraz de un litro que contenga 900 ml de agua caliente, agitar, dejar enfriar y enrasar a un litro. En vez de ácido sórbico puede emplearse 26,8 g de sorbato potásico.
22.4 Procedimiento.
Tomar 0,5 ml del destilado de la acidez volátil, llevar a la cubeta de 1 cm de espesor interno, añadir 1,5 ml de la solución a), dejar la cubeta al aire durante unos minutos y medir después la absorbancia en el espectrofotómetro a 256 nm. Graduar el espectrofotómetro con 0,5 ml de la solución b) y 1.5 ml de la solución a).
22.5 Cálculo.
Un gramo de ácido sórbico corresponde a 8,92 ml de solución normal y a 0,438 g de ácido sulfúrico. Para todo vino adicionado con 200 mg de ácido sórbico por litro la corrección sustractiva que corresponde hacer a la acidez volátil es de 1,7 meq o 0,088 g del ácido sulfúrico o 0,107 g de ácido acético por litro de vino.
22.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. All, 3.
23. ACIDEZ FIJA
23.1 Principio.
Se considera convencionalmente como acidez fija la diferencia entre acidez total y acidez volátil, ambas expresadas en ácido tártrico.
No es necesario hacer las correcciones por eventual presencia de anhídrido sulfuroso combinado y libre.
24(a) ÁCIDO TÁRTRICO TOTAL
(Método del racemato de calcio)
24(a).1 Principio.
Adición al vino de ácido tártrico levógiro que en unión con el ácido tártrico dextrógiro del vino y en presencia de solución de calcio, forman en condiciones determinadas racemato de cal insoluble.
En el método adoptado se determinan las condiciones de precipitación del racemato de calcio, fijando la solubilidad del tartrato levógiro de calcio (que también se forma) y las del racemato de calcio en función del pH y de la riqueza en iones de calcio. Con estos datos se ha fijado un pH conveniente para que precipite el racemato de calcio y se solubilice el tartrato levógiro.
24(a).2 Material y aparatos.
24(a).2.1 Vaso cilíndrico de 600 ml.
24(a).2.2 Filtros de porosidades números 4 y 2.
24(a).2.3 Kitasato y trompa de agua para vacío.
24(a).2.4 Material accesorio para una volumetría.
24(a).3 Reactivos.
24(a).3.1 Solución de acetato de cal de 10 g de calcio por litro. Mezclar en matraz aforado de 1.000 ml 25 g de carbonato cálcico con 40 ml de ácido acético cristalizable y enrasar con agua destilada hasta 1.000 ml.
24(a).3.2 Racemato de calcio cristalizado. En un vaso, introducir 20 ml de una solución de ácido tártrico de 5 g por litro, 100 ml de agua destilada, 20 ml de una solución de tartrato levógiro de amonio de 6,126 g por litro y 6 ml de la solución de acetato de calcio.
Dejar precipitar durante dos horas. Recoger el precipitado sobre un filtro de porosidad número 4, lavando y arrastrando cuatro veces. Secar en estufa a 70° hasta peso constante. Se obtienen unos 340 mg de racemato de calcio cristalizado.
Conservar en frasco tapado.
24(a).3.3 Licor de precipitación. Introducir en matraz aforado de 1.000 ml 150 g de tartrato de amonio levógiro, 8,8 ml de solución de calcio de 10 g/l y enrasar con agua destilada.
Por ser el racemato de calcio ligeramente soluble en este licor, conviene saturarle antes de su empleo, añadiendo 5 g de racemato de calcio a un litro de este licor. Agitar durante 12 horas y filtrar.
24(a).4 Procedimiento.
24(a).4.1 Precipitación del racemato.‒En un vaso cilíndrico de 600 ml introducir 500 ml de licor de precipitación y 10 ml de vino. Mezclar y dejar precipitar durante 24 horas.
24(a).4.2 Valoración gravimétrica.‒Filtrar sobre filtro poroso número 2, tarado y puesto sobre el Kitasato, recogiendo el precipitado. Lavar el vaso en el que se ha efectuado la precipitación y arrastrar las últimas partículas del precipitado con el líquido ya filtrado.
Secar en estufa a 70° hasta peso constante.
24(a).5 Cálculo.
Calcular el ácido tártrico expresado en g/l de vino o en g de tartrato ácido de potasio.
Acido tártrico = 28,84 X P g/l
Acido tártrico = 36,15 X P g de tartrato ácido de potasio.
P = peso en g del racemato de calcio, cristalizado con cuatro moléculas de agua.
24(a).6 Referencia.
1. Jaulmes, P.; Brum, S.; Cabanis, J. «Comunicación número 304 de Métodos de Análisis de Vinos». O. I. V., 1969.
24(b). ÁCIDO TÁRTRICO TOTAL
(Método complexométrico)
24 (b).1. Principio.
Como en 24 (a).1.
24(b).2 Material y aparatos.
24(b).2 (1, 2, 3, 4) como 24(a).2 (1, 2, 3, 4).
24(b).3 Reactivos.
24(b).3.1 Solución tampón pH = 9. Mezclar 54 g de cloruro de amonio, 350 ml de NH4(OH) en solución al 25 por 100 y completar con agua hasta un litro.
24(b).3.2 Indicador de negro eriocromo T. Mezclar 1 g de negro eriocromo T con 100 g de cloruro de sodio puro.
24(b).3.3 Solución de magnesio 0,05 M. Disolver 2,25 g de óxido de magnesio puro (para asegurar que no esté carbonatado se recomienda calcinar 5 g de óxido de magnesio «puro» al rojo en horno eléctrico durante 30 minutos, enfriar y pesar 2,25 g). Hacer la disolución en cantidad necesaria de ácido clorhídrico diluido (10 por 100) y luego se enrasa a un litro.
Valorar esta solución con otra 0,05 M de complexona III en presencia de negro de eriocromo.
24(b).3.4 Solución de complexona III 0,05 M. Disolver 18,61 g de complexona III (sal disódica bihidratada del ácido diamino tetracético) en un litro de agua destilada.
24(b).3.5 Ácido clorhídrico diluido a 1/5 (V/v).
24(b).3.6 Na(OH) en solución N.
24(b).4 Procedimiento.
Disolver en 10 ml de ácido clorhídrico diluido el precipitado de racemato recogido sobre el crisol filtrante. Lavar el crisol filtrante con unos 50 ml de agua destilada. Añadir un volumen de solución Na(OH) N/l suficiente para neutralizar la solución (unos 20-22 ml). Añadir después 5 ml de solución tampón pH = 9,5 ml de sal de magnesio 0,05 M y 50 mg de indicador negro eriocromo T. Valorar con la complexona III 0,05. Molar.
24(b).5 Cálculo.
Ácido tártrico en g/l de vino = (V ‒ 5) 3,75.
Ácido tártrico expresado en g/l de tartrato ácido de potasio = (V ‒ 5) 4,70.
V = volumen en ml de complexona III 0,05 M.
La expresión de los resultados serán en tartrato ácido de potasio y con una aproximación de 0,5 miliequivalentes por litro, o de 0,05 g/l.
25. ÁCIDO LÁCTICO
25.1 Principio.
Aislamiento del ácido láctico mediante columna de resinas cambiadoras de aniones, oxidación a acetaldehído y valoración colorimétrica por la reacción coloreada del acetaldehído con la piperidina.
25.2 Material y aparatos.
25.2.1 Espectrofotómetro y accesorios.
25.2.2 Tubo de vidrio o bureta de 10-11 mm de diámetro y 30 cm de longitud con llave en la parte inferior.
25.2.3 Lana de vidrio.
25.2.4 Resina cambiadora de aniones.
25.3 Reactivos.
25.3.1 Solución 0,1 M de sulfato cérico (cerio IV) en ácido sulfúrico 0,7 N. Disolver en frío 40,431 g de sulfato cérico, Ce (SO4)2 4H2O en 350 ml de ácido sulfúrico 2 N exactamente valorado. No calentar, pues se formaría un óxido cérico insoluble. Llevar la solución a 1.000 ml con agua destilada.
25.3.2 Solución valorada de Na(OH) 2,5 N.
25.3.3 Solución de acetato de sodio al 27 por 100. Disolver 270 g de acetato de sodio desecado en agua y completar 1.000 ml.
25.3.4 Solución valorada de SO4H2 2 N.
25.3.5 Solución de nitroprusiato de sodio al 2 por 100. Disolver 2 g de nitroprusiato de sodio pulverizado en agua, después completar a 100 ml. Conservar tapado en la oscuridad. La solución no debe tener más de ocho días.
25.3.6 Solución de piperidina al 10 por 100. Llevar a un matraz de 100 ml, 10 ml de piperidina y enrasar a 100 ml con agua destilada.
25.4 Procedimiento.
25.4.1 Preparación de la columna de resinas aniónicas. Colocar lana de vidrio en el fondo del tubo o bureta 25.2.2, formando tapón de 2-3 mm de altura aproximadamente, añadir agua hasta una altura de unos 5 mm sobre el tapón de lana de vidrio. Añadir después la resina cambiadora de aniones, que estará conservada en ácido acético glacial al 30 por 100, agitar esta suspensión y añadir rápidamente un volumen de unos 10 mililitros. Dejar sedimentar y después depositar en la superficie un tapón de lana de vidrio que se conduce a través del líquido sin resinas con una varilla de vidrio.
La resina cambiadora de aniones no sirve más que para una sola vez.
25.4.2 Aislamiento del ácido láctico. Abrir la llave inferior de la columna y dar salida al ácido acético diluido hasta unos 2-3 ml por encima del tapón de lana superior. Añadir unos 10 mililitros de ácido acético al 0,5 por 100, vaciar hasta igual altura y repetir estos lavados cuatro veces.
Después del último lavado, y con llave cerrada, adicionar 10 ml del vino y señalar la altura alcanzada, con lápiz graso. Dejar salir goteando el vino a razón de 1-1,5 gotas por segundo (25-30 ml por minuto). Vaciar hasta el nivel del tapón de lana de vidrio superior. Llenar de nuevo la columna con ácido acético al 0,5 por 100 hasta la señal de lápiz graso. Dejar salir a la misma velocidad que la vez anterior y lavar después en la misma forma siete veces, cada vez con 10 ml de agua destilada.
Al terminar el último lavado, cerrar la llave cuando el nivel del líquido se encuentre un poco más arriba del tapón de lana de vidrio superior.
Colocar un matraz receptor aforado de 100 ml. Eluir los ácidos fijados en el cambiador de aniones mediante adiciones de solución de sulfito de sodio 0,5 M, hasta la señal trazada en el tubo.
Resulta práctico, para realizar esta operación, colocar un frasco con la solución de sulfito unido por el cuello a la columna mediante un manguito de caucho y con unas pinzas para regular la caída del líquido en la bureta o tubo. Puestos así en comunicación los dos aparatos (frasco y bureta o tubo), abrir las pinzas del caucho de unión y la llave inferior, dejando caer el líquido a la bureta hasta unos 10 cm de altura, y sin que queden huecos vacíos. Se regula la salida del líquido a la proporción de 2-3 gotas por segundo, para llenar el matraz receptor hasta el enrase.
25.4.3 Valoración del ácido láctico. Introducir 10 ml del eluido en un tubo de ensayo de paredes que no sean gruesas y de unos 50 ml de capacidad con tapón. Añadir 10 ml del reactivo de sulfato cérico. Agitar e introducir el tubo de ensayo en un termostato a 65 °C durante 10 minutos exactamente. Inmediatamente después de la inmersión en el líquido, destapar durante unos segundos, para dar salida al aire dilatado. Cerrar rápidamente con el tapón para evitar pérdidas del acetaldehído formado. Después de 10 minutos, retirar y enfriar el tubo con agua corriente hasta la temperatura de 20 °C.
Añadir 5 ml de la solución de hidróxido de sodio 2,5 N, mezclar bien y filtrar.
Tomar 15 ml del filtrado y verterlos en probeta de tapón esmerilado de 50 ml de capacidad, que contenga 5 ml de solución de acetato de sodio al 27 por 100 y 2 ml de ácido sulfúrico 2 N. Añadir además 5 ml de solución de nitroprusiato de sodio y mezclar bien. Añadir después 5 ml de solución de piperidina, mezclar rápidamente, introducir en seguida en la cubeta del espectrofotómetro de 10 mm de espesor. Medir la coloración producida, que varía, de verde a violeta, con relación al aire a la longitud de onda de 570 nm. Esta coloración aumenta al principio, pero disminuye en seguida rápidamente. Tomar como valor definitivo el valor máximo de la lectura de absorbancia.
Si el eluido es muy rico en ácido láctico y la densidad óptica es muy elevada, diluir el eluido con solución de sulfato de sodio al 7,1 por 100.
25.4.4 Curva patrón. Tomar 10 ml de una solución de ácido láctico N exacta, y añadir 10 ml de solución valorada de hidróxido de sodio N y completar a un litro con solución de sulfato de sodio al 7,1 por 100. Tomar 5, 10, 15, 20 y 25 ml, respectivamente, e introducir cada alícuota en un matraz aforado de 100 mililitros. Enrasar con solución de sulfato de sodio al 7,1 por 100. Mezclar bien, tomar 10 ml de las soluciones así obtenidas y determinar los valores de las absorbancias respectivas.
Las diferentes soluciones corresponden a eluido del vino conteniendo 0,45, 0,90, 1,35, 1,80 y 2,25 g/1 de ácido láctico.
La representación gráfica de las absorbancias de estas soluciones en función de la riqueza en ácido láctico, es una recta.
25.5 Cálculo.
Llevar los valores de absorbancia obtenidos a la curva patrón y deducir los correspondientes contenidos en ácido láctico expresados en g/l.
25.6 Observaciones.
Si el vino tiene más de 250 mg por litro de sulfuroso total, el resultado puede ser erróneo, por la presencia de ácido aldehído sulfuroso. En determinaciones de precisión, conviene realizar una corrección en la forma siguiente:
Tomar 15 ml de eluido y llevar a probeta de tapón esmerilado, en la que se habrá añadido 5 ml de acetato de sodio al 27 por 100 y 2 ml de SO4H2 1,55 N (77,5 ml de SO4H2 2 N diluidos a 100 ml con agua). Añadir 5 ml de nitroprusiato de sodio al 2 por 100 y 5 ml de piperidina al 10 por 100. Mezclar y hacer la determinación espectrofotométrica a 570 nm que da el factor de corrección:
L = L' ‒ C × 0,25
L = contenido real de ácido láctico expresado en g/l.
L' = contenido aparente de ácido láctico expresado en g/I.
C = factor de corrección en g/l.
25.7 Referencia.
1. Rebelein, M. «Deutsche Lebens-mittel Rudshan», 2: 36-41, 1961, y 59: 131, 1963.
26. ÁCIDO CÍTRICO
26.1 Principio.
El ácido cítrico es fijado junto con los otros ácidos del vino por una resina cambiadora de aniones. Después se procede a la elución, la que se realiza fraccionadamente y separando el ácido citromálico que es causa de error en la valoración del ácido cítrico.
El ácido cítrico es transformado por oxidación cuidadosa en acetona, la que se separa por destilación. El etanal arrastrado se oxida a ácido acético y se valora sola la acetona por iodometría.
26.2 Material y aparatos.
26.2.1 Columna cambiadora de aniones. En una bureta de llave de 25 ml de capacidad, colocar un tapón de lana de vidrio en el fondo y verter 20 ml de resina Dowex 1 x 2.
Someter la resina a dos ciclos completos de regeneración con pasos alternos de solución N de ácido clorhídrico e hidróxido de sodio. Lavar después con 50 ml de agua destilada, agitando la resina cuando los primeros ml de agua pasan por la columna para despegarla del fondo de la bureta.
Saturar la resina de iones acetato, mediante el paso de 250 mililitros de una solución 4 N de ácido acético. Lavar luego con 100 ml de agua destilada.
26.2.2 Aparato para la oxidación. Aparato compuesto de matraz de cuello largo, atravesado lateralmente por el tubo afilado de embudo con llave, y en comunicación con refrigerante, según dimensiones y forma del diseño (fig. 26.1.).
26.3 Reactivos.
26.3.1 Resina Dowex 1 x 2 (50-100 mallas).
26.3.2 Solución 4 N de ácido acético.
26.3.3 Solución 2,5 N de ácido acético.
26.3.4 Solución 2 N de hidróxido de sodio.
26.3.5 Ácido sulfúrico diluido al 1/5 (V/V).
26.3.6 Solución tampón pH = 3,2-3,4. En un matraz aforado de 1 l introducir 150 g de fosfato monopotásico y 5 ml de ácido fosfórico puro (d = 1,689) y enrasar con agua destilada.
26.3.7 Solución de sulfato de manganeso de 50 g/l.
26.3.8 Piedra pómez.
26.3.9 Solución 0,05 N de permanganato potásico.
26.3.10 Ácido sulfúrico diluido a 1 (V/V).
26.3.11 Solución 2 N de permanganato potásico.
26.3.12 Solución de sulfato ferroso de 40 g por 100.
26.3.13 Solución 5 N de hidróxido de sodio.
26.3.14 Solución 0,02 N de iodo.
26.3.15 Solución 0,02 N. de tiosulfato sódico.
26.3.16 Engrudo de almidón.
26.4 Procedimiento.
26.4.1 Separación de los ácidos cítrico y citromálico. Hacer pasar 25 ml del vino a través de la columna cambiadora de aniones Dowex 1 x 2 en forma acética a razón de 3 ml cada dos minutos. Lavar la columna con 20 ml de agua destilada en tres veces. Eluir los ácidos con 200 ml de solución 2,5 N de ácido acético que se hace pasar por la columna a la misma velocidad. En esta fracción de eluido se encuentra, entre otros ácidos, el ácido citromálico que interesa eliminar.
Eluir los ácidos cítricos y tártricos haciendo pasar a través de la columna 100 ml de una solución 2 N de hidróxido de sodio y recoger el eluido en el matraz del aparato de oxidación y destilación descrito.
26.4.2 Oxidación. En el matraz que contiene ya el eluido anterior, añadir ácido sulfúrico diluido al 1/5 (unos 20 ml para llevar el pH a 3,2-3,8, 25 ml de solución tampón pH = 3,2-3,4 1 ml de solución de sulfato de manganeso y algunos gramos piedra pómez. Tapar y conectar el matraz con el refrigerante, calentando hasta ebullición. Separar los primeros 50 ml destilados.
Colocar solución 0,05 N de permanganato de potasa en el embudo lateral de llave, y verter en el matraz a razón de una gota por segundo, cayendo en el eluido en ebullición. El destilado se recoge en frasco de 500 ml de tapón esmerilado que contiene unos ml de agua. Continuar la oxidación hasta que la coloración parda del líquido acuse un exceso de permanganato.
26.4.3 Separación de la acetona. Si el volumen del destilado es inferior a 90 ml, completar con agua hasta este volumen: añadir después 4,5 ml de ácido sulfúrico al 1/3 y 5 ml de una solución 2 N de permanganato potásico.
Si el volumen del destilado excede de los 90 ml, completar a 180 ml con agua destilada y utilizar la dosis doble de reactivos.
En estas condiciones (medio 0,5 N en ácido sulfúrico y 0,1 N en permanganato de potasio), el etanol es oxidado a ácido acético y la acetona permanece intacta.
Tapar el frasco, dejar en reposo durante 45 minutos a la temperatura ambiente. Al cabo de este tiempo se destruye el exceso de permanganato adicionando solución de sulfato ferroso.
Destilar después y recoger 50 ml aproximadamente de destilado en un frasco de tapón esmerilado que contenga 5 ml de solución 5 N de hidróxido sódico.
26.4.4 Valoración de la acetona. Añadir al contenido del frasco 25 ml de solución de iodo 0,02 N, si la dosis de ácido cítrico es superior a 0,5-0,6 g/l será necesario añadir más solución de iodo, hasta que quede color amarillento que indicará este exceso. Duplicar o triplicar las dosis de iodo hasta llegar a este punto, pero si la riqueza en ácido cítrico es mayor de 1,5 g, comenzar otra vez el análisis con sólo 10 ml de vino.
Dejar en contacto el iodo añadido durante 20 minutos permaneciendo el frasco tapado. Añadir 8 ml de ácido sulfúrico al 1/5 y valorar el exceso de iodo con tiosulfato 0,02 N en presencia de almidón.
Hacer una valoración en blanco en las mismas condiciones sustituyendo los 50 ml de destilado por 50 ml de agua destilada.
26.5 Cálculo.
Calcular el contenido en ácido cítrico expresado en g/l.

V = volumen en ml de la muestra de vino.
V' = volumen en ml de la solución de tiosulfato 0,02 N utilizado en la valoración en blanco.
V" = volumen en ml de la solución de tiosulfato 0,02 N utilizado en la valoración del iodo en exceso.
26.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A29, 1-3, 1969.
27. SULFATOS
27.1 Principio.
Precipitación del sulfato de bario en el vino, previamente desprovisto de anhídrido sulfuroso por ebullición al abrigo del aire.
27.2 Material y aparatos.
27.2.1 Matraz cónico (erlenmeyer) de 500 ml, provisto de ampolla con llave de 100 ml y de desprendimiento de vapor, atravesando ambos el tapón. El matraz se coloca sobre disco metálico de 15 cm de diámetro con abertura circular de 8 cm de diámetro.
27.2.2 Vaso cilíndrico de 400 ml.
27.2.3 Baño de agua o plancha caliente a 60 °C.
27.3 Reactivos.
27.3.1 Ácido clorhídrico 2 N.
27.3.2 Cloruro de bario, 100 g de BaCl2 ∙ 2H2O en 1.000 ml de agua.
27.3.3 Ácido clorhídrico puro.
27.4 Procedimiento.
27.4.1 Eliminación del anhídrido sulfuroso del vino. Introducir en el erlenmeyer del aparato descrito 50 ml de agua destilada y 1 ml de ácido clorhídrido puro. Hervir esta solución para eliminar el aire del aparato. Introducir 100 ml del vino por la ampolla de llave evitando que la ebullición se pare durante esta adición. Continuar la ebullición hasta reducir el volumen del líquido a unos 100 ml, eliminando así todo el SO2.
27.4.2 Precipitación de sulfato de bario. Llevar el residuo de la evaporación anterior a un vaso de 400 ml, enjuagando el matraz con la cantidad de agua suficiente para llevar el volumen total a 200 ml. Añadir 5 ml de ácido clorhídrico 2 N y llevar a ebullición. Adicionar gota a gota 10 ml de solución de cloruro de bario, con la precaución de no parar la ebullición y dejar reposar en caliente, colocando el vaso sobre placa a 60° durante cuatro horas y en baño de agua hirviendo durante dos horas. En el caso de pequeñas cantidades de precipitado, se recomienda un segundo reposo en frío durante 12 horas. Filtrar por filtro sin cenizas, lavar el vaso y precipitado con agua caliente hasta que no dé reacción con NO3Ag, llevar el filtro cuidadosamente plegado a crisol de porcelana tarado, desecar, calcinar y, después de enfriar en un desecador, pesar.
27.5 Cálculo.
Calcular el contenido de sulfatos expresado en meq/l o en g/l de sulfato potásico con una aproximación de ± 0,05 g/l.
Sulfato = 85,68 P meq/I.
Sulfato = 7,465 P g/1 de sulfato potásico.
P = peso de las cenizas en g.
27.6 Referencias.
1. Deibner, P.; Bernard, P. «Industries Alimentaires et Agricoles., 71: 1, 23 y 5, 427, 1954
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A14, 1-4.
28(a). ANHÍDRIDO SULFUROSO
(Método Paul)
28(a).1 Principio.
Liberación de sulfuroso «libre» por acidificación del vino, arrastre por corriente de aire, oxidación por barboteo en agua oxigenada neutra y valoración con sosa del ácido sulfúrico formado. Liberación por ebullición moderada del sulfuroso «combinado», que queda en el vino después de la extracción del sulfuroso «libre», y análogo tratamiento que en la determinación de sulfuroso «libre». El sulfuroso «total» es la suma del sulfuroso «libre» y el sulfuroso «combinado». Puede determinarse acidificando el vino y calentando y procediendo como en los dos casos anteriores.
28(a).2 Material y aparatos.
Utilizar el aparato representado en la figura 28(a).I.
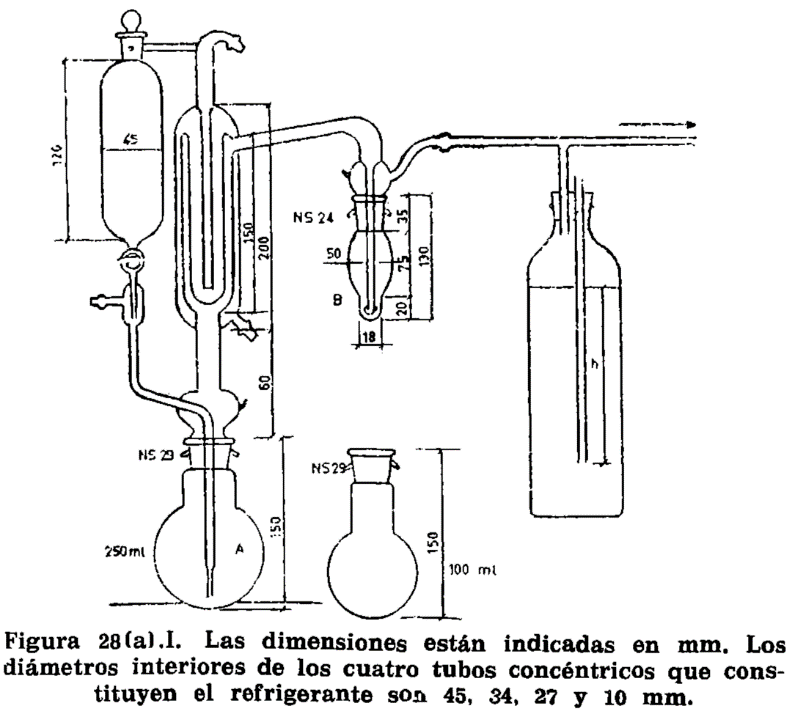
28(a).2.1 Matraz de 100 ml o de 250 ml.
28(a).2.2 Tubo barbotador provisto de bola hueca en un extremo con uno: 20 orificios de 0,2 mm de diámetro alrededor del circule máxime horizontal.
28(a).2.3 Refrigerante que condense vapores y deje pasar el gas en recorrido seguro.
28(a).2.4 Frasco con agua y con tapón atravesado por el tubo que comunica con el barbotador para hacer vacío, y otro tubo sumergido en el agua para acusar la intensidad del vacío por la depresión de la columna de agua en el interior del tubo, depresión que debe mantenerse entre 20-30 cm.
El gasto de vapor será de 40 l/h.
28(a).2.5 Baño de agua regulable a 10 °C.
28(a).3 Reactivos.
28(a).3.1 Ácido fosfórico al 25 por 100 (P/V).
28(a).3.2 Agua oxigenada de 0,3 volúmenes por 100.
28(a).3.3 Hidróxido sódico N/100.
28(a).3.4 Indicador. Mezclar 100 mg de rojo de metilo, 50 mg da azul de metileno y 100 ml de alcohol de 50°.
28(a).4 Procedimiento.
28(a).4.1 Análisis del anhídrido sulfuroso libre: En el matraz de 100 ml del aparato poner 10 ml del vino y añadir 5 ml de ácido fosfórico al 25 por 100, colocando acto seguido el matraz en su sitio. Si la riqueza en SO2 del vino es pequeña, se emplea el matraz de 250 ml y se ponen 20-25 ml de vino.
Sumergir el matraz en un baño de agua a 10 °C.
Colocar 2-3 ml de agua oxigenada de 0,3 volúmenes y dos gotas del reactivo indicador en el barbotador y neutralizar el agua oxigenada con Na(OH) N/100.
Adaptar el barbotador al aparato y hacer barbotar el aire (o nitrógeno) durante 12-15 minutos. Arrastrar el anhídrido sulfuroso libre y después oxidar a SO4H2. Retirar el barbotador y valorar el ácido formado con solución de Na(OH) N/100.
28(a).4.2 Análisis del anhídrido sulfuroso combinado. Después de terminar la valoración del anhídrido sulfuroso libre, colocar en el barbotador ya limpio, los 2-3 ml de agua oxigenada neutralizada y con las dos gotas del indicador. Calentar con llama pequeña hasta llevar a la ebullición el vino que quedó en el matraz despojado del anhídrido sulfuroso libre por la determinación anterior. Aplicar el fuego directo sobre el fondo del matraz, que descansará sobre chapa metálica perforada con orificio de 30 mm de diámetro, para evitar la pirogenación de las materias extractivas del vino sobre las paredes del matraz. Mantener el paso del aire o nitrógeno durante la ebullición, que durará 12-15 minutos, tiempo que se considera suficiente para arrastrar todo el sulfuroso combinado, y después oxidarlo.
Continuar como en 28(a).4.1 a partir de «Colocar 2-3 ml de agua oxigenada...».
28(a).4.3 Anhídrido sulfuroso total. Puede determinarse por la suma anhídrido sulfuroso libre más anhídrido sulfuroso combinado, pero también puede determinarse directamente, actuando desde un principio con corriente de aire y con calor.
28(a).5 Cálculo.
Calcular el contenido en anhídrido sulfuroso expresado en mg/l con una aproximación de 10 mg/l.
Anhídrido sulfuroso = 32 V mg/l.
V = volumen en ml de NaOH N/100.
28(a).6 Observaciones.
Para vino sensiblemente picado (con más de 2 g/l de acidez acética) puede arrastrarse con el anhídrido sulfuroso, ácidos volátiles, especialmente cuando la corriente del aire es intensa.
Corregir determinando la acidez volátil presente en el líquido del barbotador, operando de la siguiente forma:
Después de la neutralización con la sosa al valorar la acidez, añadir un cristal de ácido tártrico y llevar el contenido del barbotador al aparato de determinación de la acidez volátil por destilación en corriente de vapor. Restar después del resultado anterior la parte de acidez correspondiente a los ácidos volátiles.
28(a).7 Referencias.
1. Paul, F. «Mitteilungen Klosternenberg, A: 8, 21, 1958.
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A17, 1-8, 1969.
28(b). ANHÍDRIDO SULFUROSO
(Método Ripper doble)
28(b).1 Principio.
Valoración iodométrica del SO2, operando directamente con el vino.
Para evitar el error introducido por la recombinación del sulfuroso liberado de su combinación con los aldehidos, durante la valoración volumétrica, se hace una segunda valoración, después de nueva liberación del SO2 recombinado.
28(b).2 Material y aparatos.
28(b).2.1 Erlenmeyer de 500 ml y material para una volumetría.
28(b).3 Reactivos.
28(b).3.1 Hidróxido sódico 4 N.
28(b).3.2 Ácido sulfúrico al 1/10 en volumen (180 g/1).
28(b).3.3 Engrudo de almidón preparado de la siguiente forma:
Pesar 2,5 de almidón soluble y triturarlo en un mortero junto con 10 mg de ioduro mercúrico. Llevar a un vaso y añadir un volumen de agua suficiente para poder hervir la suspensión con fluidez. Llevar después ésta a matraz con 1 litro de agua en ebullición, que se mantiene durante 10 minutos. El líquido obtenido estará limpio y si es necesario se filtra.
28(b).3.4 Solución de iodo N/20.
28(b).3.5 Tiosulfato sódico N/100.
28(b).3.6 Versenato de sodio (complexona III).
28(b).4 Procedimiento.
Colocar en el erlenmeyer de 500 ml, 50 ml del vino, 3 ml de SO4H2 al 1/10, 5 ml de engrudo de almidón y 30 mg de versenato sódico (con el fin de evitar la oxidación del SO2 libre durante la valoración).
Homogeneizar la solución y añadir con bureta solución de I N/20 hasta coloración azul del almidón, que primero será fugaz, y netamente persistente después, durante 10-15 segundos, considerándola entonces aceptable. Sea V el volumen en ml del iodo empleado.
Añadir 8 ml de solución 4 N de hidróxido sódico, agitar una sola vez y dejar en contacto durante 5 minutos. Después verter, de un solo golpe y agitando enérgicamente, 10 ml de SO4H2 al 1/10.
Se valora inmediatamente con iodo N/20 el SO2 liberado sea V'' el volumen en ml de solución de iodo empleado.
Se añade después 20 ml de Na(OH), 4 N, se deja en contacto durante 5 minutos después de haber agitado una sola vez. Se diluye con 200 ml de agua bien fría, agitar después enérgicamente y añadir 30 ml de SO4H2 al 1/10 y valorar con solución de iodo N/20 el SO2 liberado. Sea V" el volumen en ml de la solución de iodo empleado.
Con estas valoraciones se corrige el error que puede originarse por la recombinación de parte del SO2 ya liberado de los aldehídos, con el mismo aldehído que estará presente en el líquido que se valora.
También es posible causa de error, y especialmente cuando se trate de vinos tintos ricos en color y extractos, el consumo de iodo por estas sustancias; para corregir este error se opera de la manera siguiente:
Adicionar a 50 ml del vino, colocados en un erlenmeyer de 300 ml, un exceso de etanol para que sea capaz de combinarse con todo el SO2 libre; dicha cantidad puede ser de unos 5 ml de solución de acetaldehído, de 7 g por litro Se tapa el matraz y después de un reposo de 30 minutos, como mínimo, se añaden 3 ml de SO4H2 diluido al 1/10 y después los de solución de iodo N/20 necesarios hasta el viraje de almidón. Sea V'' el volumen en ml necesarios.
La presencia de ácido ascórbico también puede ser causa de error. Descontar el iodo correspondiente, considerando que 1 ml de iodo N/20 es consumido por 4,4 mg de ácido ascórbico.
28(b).5 Cálculo.
SO2 libre = 32 (V ‒ mg/l.
SO2 combinado = 32 (V' + V") mg/l.
SO2 total = 32 (V + V ‒ V"') mg/l.
Con aproximación de 10 mg/l.
28 (b).6 Observaciones.
Para vinos pobres en SO2, es conveniente emplear iodo más diluido, solución N/50, reemplazando entonces el coeficiente 32 por el de 12,8 en los cálculos.
En los vinos tintos es conveniente, para apreciar el viraje, iluminar con luz amarilla el fondo del erlenmeyer donde está el vino. Hay dispositivos sencillos a base de simple bombilla con filtro preparado con solución de cromato potásico, o bien con luz amarilla de lámpara de vapor de sodio.
Cuando haya un interés especial en la determinación del SO2 libre, debe dejarse la muestra en reposo durante 4 días, al abrigo del aire y a temperatura de 20 °C. El análisis se realizará también a 20 °C.
28.(b).7 Referencias.
1. Jaulmes P. y Dienziede. «Méthode Ripper double d'analyse de SO. Bol.». O. I. V., 274. 52-54, 1959.
2. Recueil des Méthodes Internationales d'Analyse des Vins O. I. V., A17, 7. 1969.
29. ÁCIDO SÓRBICO
29.1 Principio.
El ácido sórbico arrastrado por el vapor de agua (constante de volatilidad 0,59), se valora en el destilado por espectrofotometría en el ultravioleta.
29.2 Material y aparatos.
29.2.1 Espectrofotómetro ultravioleta y cubetas de cuarzo de 1 cm de espesor.
29.2.2 Matraces aforados de 20 ml y de 1 litro.
29.2.3 Pipetas de 0,5 ml y 1,5 ml.
29.3 Reactivos.
29.3.1 Solución (a) (destinada a catalizar la oxidación del ácido sulfuroso por el aire). Mezclar 0,5 g de NaHCO3 con 0,001 g de CuSO4 ∙ 5H2O y añadir agua hasta 1.000 ml.
29.3.2 Solución (b) de ácido sórbico de 20 mq por litro. Introducir 20 ml de ácido sórbico en un matraz aforado de 1 litro que contenga 900 ml de agua caliente, agitar, dejar enfriar y enrasar a 1 litro.
En vez de ácido sórbico puede emplearse 26,8 mg de sorbato potásico.
29.4 Procedimiento.
Destilar como en 21.4, 20 ml de vino adicionado de 1-2 g de ácido tártrico. Recoger 320-330 ml de destilado para conseguir la más completa extracción del ácido sórbico, que requiere unos 100 ml más que los necesarios para la acidez volátil.
Tomar 5 ml del destilado, colocar en un matraz aforado 20 ml y enrasar con la solución (a).
Llenar con esta mezcla una cubeta de cuarzo de 1 cm de espesor. Dejar la cubeta al aire durante unos minutos. Determinar la absorbancia a 256 nm. Para la prueba en blanco, sustituir los 5 ml de destilado por 5 ml de agua destilada.
Para obtener la curva patrón, determina; la absorbancia de soluciones que contengan 0,5, 1, 2,5 y 5 mg/l de ácido sórbico obtenidas diluyendo con agua destilada la solución (b). La gráfica correspondiente a estos valores es una línea recta.
29.5 Cálculo.
Calcular el ácido sórbico expresado en mg/l. Acido sórbico = 0,2 ∙ C, y mg/l.
C = contenido en mg/l de ácido sórbico determinado por comparación con curva patrón.
V = volumen en ml de destilado.
29.6 Referencias.
1. Jaulmes, P.; Mestres, R, y Mandrou, B. «Annales des Falsifications et de l'Expertise Chimique», 111, 1961.
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A30, 1-3, 1969.
30. CLORUROS
30.1 Principio.
Valoración argentimétrica después de oxidación permangánica del vino previamente defecado por barita.
30.2 Material y aparatos.
30.2.1 Matraz aforado de 200 ml.
30.2.2 Matraz de 500 ml.
30.2.3 Baño de agua.
30.3 Reactivos.
30.3.1 Solución saturada de permanganato potásico al 6,5 por 100.
30.3.2 Acido nítrico al 1/5 en volumen.
30.3.3 Solución de nitrato de plata N/10.
30.3.4 Solución de tiocianato potásico N/10.
30.3.5 Solución de alumbre de hierro y de amonio de 15 g por 100 ml o solución de nitrato férrico de 10 g por 100 ml.
30.3.6 Solución de hidróxido de bario de 50 g por litro (aproximada).
30.4 Procedimiento.
Colocar 100 ml del vino en matraz aforado de 200 ml. Neutralizar con solución de hidróxido de bario en presencia de fenolftaleína, enrasar con adición de agua destilada, agitar, filtrar por filtro de papel plisado, previamente lavado con agua tibia.
En un matraz de 500 ml colocar 100 ml de filtrado y añadir 20 ml de ácido nítrico al 1/5 y 5 ml de permanganato de potasio en solución saturada de 6,5 g por 100. Agitar y dejar reposar algunos minutos hasta desaparición del color violeta. Llevar el matraz a baño de agua y calentar sin exceso, para activar esta decoloración. Si el color persiste, se añaden unas gotas de solución de agua oxigenada. En algunos casos de vinos de intenso color, son necesarias nuevas adiciones de solución de permanganato potásico y de agua oxigenada, hasta decoloración completa.
Añadir al filtrado así tratado 10 ml de solución de nitrato de hierro al 10 por 100, 20 ml de éter y 10 ml de solución N/10 de nitrato de plata. Esta cantidad que se considera suficiente para riquezas menores de 1 g por litro de cloruros expresados en cloruro de sodio.
Valorar el exceso de nitrato de plata con solución N/10 de tiocianato de potasio, hasta viraje al color teja pálido del líquido, que deba durar por lo menos cinco segundos.
30.5 Cálculo.
Calcular el contenido en cloruros expresado en meq/l de cloruro de sodio con aproximación de 0,05 g/l.
Cloruros = 2 (10 ‒ V) meq/l.
Cloruros = 0,1169 (10 ‒ V) g/l de cloruro de sodio.
V = volumen en ml de solución N/10 de tiocianato de potasio.
30.6 Referencia.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., A15, 1-2, 1989.
31. ÁCIDO BENZOICO
31.1 Principio.
Extracción por el éter del ácido benzoico presente en el vino. Tratamiento del extracto por la mezcla nitro-sulfúrica y formación del ácido 3-5 dinitrobenzoico, que es extraído con éter y después recogido por la mezcla acetona-alcohol. La adición de sosa da origen a una coloración violeta. Esta reacción es sensible, específica y cuantitativa, siempre que se opere en condiciones que aseguren la transformación del ácido benzoico en compuesto coloreado medible en el espectrofotómetro a 570 nm.
31.2 Material y aparatos.
31.2.1 Espectrofotómetro que permita lecturas a 570 mμ.
31.2.2 Ampolla de decantación de 250 ml.
31.2.3 Matraces de 100 ml aforados.
31.2.4 Cápsulas.
31.2.5 Baño de agua.
31.2.6 Matraces aforados de 50 ml.
31.2.7 Erlenmeyer de 50 ml.
31.3 Reactivos.
31.3.1 Éter sulfúrico.
31.3.2 Mezcla acetona-alcohol: Mezclar 1.000 ml de acetona pura con 1.000 ml de alcohol absoluto.
31.3.3 Mezclar nitro-sulfúrico: Añadir a 10 g de NO3K puro para análisis, 100 ml de SO4H2.
31.3.4 Sosa alcohólica aI 5 por 100. Añadir a 5 g NaOH puro (sin CO3Na2) 50 ml de agua. Después de fría la mezcla, se añade alcohol de 95° hasta 100 ml.
La solución debe quedar limpia.
31.4 Procedimiento.
Vinos secos o con riqueza en azúcar de 100 g/l.
Añadir a 10 ml de vino puestos en ampolla de decantación 2 ml de ácido sulfúrico al 25 por 100 y 25 ml de éter, dos veces seguidas. Decantar y filtrar el éter, por papel seco, llevarlo a matraz aforado de 50 ml y enrasar.
Poner 20 ml de la solución etérea en erlenmeyer de 50 ml, evaporar aspirando los vapores mediante vacío, permaneciendo el erlenmeyer sumergido en baño de agua fría. Añadir al residuo 5 ml de la mezcla nitrosulfúrica cuidando de empapar bien todo el residuo. Llevar después al baño de agua hirviendo durante 35 minutos y agitando de cuando en cuando, sobre todo al principio.
Enfriar en agua fría, añadir 5 ml de agua, seguir enfriando y verter el contenido en una ampolla de decantación de 250 ml. Enjuagar el erlenmeyer con 5 ml de agua, volver a verter el contenido en la ampolla y añadir 30 ml de éter. Agitar cinco minutos. Eliminar la fase acuosa después de cuidadosa decantación y lavar el éter con 5 ml de agua Decantar con cuidado (un lavado más prolongado originaría pérdidas de dinitrobenzoico en la fase acuosa, por lo que es preferible decantar bien, que volver a lavar).
Filtrar el éter por filtro revestido con sulfato sódico anhidro, lavar el filtro en seguida con 10 ml de éter. Evaporar el filtrado en una pequeña cápsula a 35°, arrastrar con tres adiciones de 5 ml de acetona-alcohol, cada vez (previamente enfriado en nevera). Recoger todos estos líquidos en un tubo de ensayo de tapón esmerilado y aforado a 15 ml.
En un tubo de ensayo de tapón esmerilado y con trazo de aforo a 15 ml, poner de 0,5 a 5 ml de la anterior solución. Este volumen debe variar con la riqueza supuesta, para obtener la reacción coloreada con el ácido dinitrobenzoico originado por 0,1 a 0,5 mg de ácido benzoico. El método es válido entre 0,03 y 1 mg, si se utiliza aparato capaz de medir absorbancias superiores a 1,5.
Diluir el volumen de líquido puesto en el tubo, con la mezcla acetona-alcohol frías, hasta 14,7 ml y añadir después 0,3 ml de sosa al 5 por 100, agitar y sumergir en hielo fundente.
Hacer la determinación en cubeta de 1/2-1 cm a 570-580 nm después de 30 minutos y antes de 70 minutos a partir del momento de la mezcla.
Utilizar como solución compensadora para comparación una mezcla de 14,5 de acetona alcohol con 0,3 ml de sosa al 5 por 100.
Para obtener la curva patrón, preparar una solución valorada de 50 mg de ácido benzoico puro en 100 ml de éter. Diluir esta solución a 1/10 con éter y tratar 10 ml de esta solución de 50 mg por litro, como se ha indicado antes para la muestra.
La densidad de óptica varía linealmente con la cantidad de ácido benzoico.
31.5 Cálculo.
Llevar la absorbancia obtenida a la curva patrón y expresar correspondiente contenido de ácido benzoico en mg/l.
31.6 Referencia.
1. Spanyar, E.; Karel, E, y Kiszel, M. «Zeitschrift für Lebenmittel Untersuchung und Forschung», 1958.
32. BROMO TOTAL
32.1 Principio.
Saponificación cuidadosa, para evitar pérdidas, de los ésteres del ácido brómico, eventualmente presentes en el vino, transformación del rojo fenol en azul de bromofenol, y valoración colorimétrica o espectrofotométrica.
32.2 Material y aparatos.
32.2.1 Cápsula de sílice de 7 cm de diámetro.
32.2.2 Baño de agua.
32.2.3 Horno eléctrico, recomendable con regulador automático, para regular a 525º.
32.2.4 Espectrofotómetro o colorímetro capaces para lectura a 584 nm.
32.3 Reactivas.
32.3.1 Solución de Na (OH) al 50 por 100.
32.3.2 Lechada de cal 4 N (224 g de CaO por litro).
32.3.3 Solución de rojo fenol. Mezclar 0,24 g de rojo fenol y 24 ml de NaOH N/10. Añadir agua destilada hasta 1.000 ml.
32.3.4 Solución tampón pH = 4,65. Mezclar 500 ml de ácido acético 2 N, 250 ml de sosa cáustica 2 N y 250 ml de agua destilada.
32.3.5 Solución oxidante. A 2 g de cloramina T añadir agua destilada hasta un litro.
Preparar esta solución 48 horas antes de su empleo y sólo será válida hasta quince días.
32.3.6 Solución reductora. Añadir a 25 g de tiosulfato de sodio agua destilada hasta un litro.
32.3.7 Solución valorada de bromuro de potasio. Añadir a 1,489 g de bromuro de potasio agua destilada hasta un litro.
Esta solución contiene 1 g de bromuro por litro. En el momento de su empleo se hacen dos diluciones sucesivas a 1/10 y se tendrá la solución a emplear que contiene 10 microgramos de bromo por ml.
32.4 Procedimiento.
Adicionar 0,5 ml de NaOH al 50 por 100 y 1 ml de lechada de cal 4 N a 50 ml del vino colocados en una cápsula de cuarzo de 7 cm de diámetro. Esta adición proporciona un soporte infusible que permite una buena combustión del carbón. Si el vino es muy ácido conviene añadir antes sosa para llevarlo a pH 10 por lo menos.
Si el vino contiene compuestos orgánicos de bromo o antisépticos bromados, dejar por lo menos 24 horas la muestra de vino alcalinizada, tapando la cápsula con un vidrio de reloj, con el fin de saturar los ésteres del ácido bromacético, que son muy volátiles.
Evaporar el líquido de la cápsula en baño de agua, calentar el residuo con pequeña llama o en baño de arena para eliminar las últimas trazas de agua. Incinerar hasta cenizas blancas en el horno eléctrico regulado a 525 °C, sin pasar de esta tempera-tura para evitar pérdidas por sublimación.
Para facilitar la obtención de cenizas blancas, retirar la cápsula después de 15 minutos de incineración, dejar enfriar y humedecer las cenizas aún grisáceas con unas gotas de agua, evaporar y volver al horno, repitiendo esta operación las veces necesarias hasta conseguir cenizas blanca...
Añadir a las cenizas 5 ml de agua hirviendo y remover con una varilla, llevar a pH 4,6-4,5 con ácido sulfúrico a 1/10 y ajustar con ácido sulfúrico a 1/100 y completar con agua hasta 10 ml.
Para calcular la cantidad de agua a añadir, tomar el volumen final igual a 10,2 ml, por considerar el aumento de volumen agua más ácido sulfúrico, equivalente a 0,2 ml, que habrá sido arrastrado en la precipitación de los 334 mg de sulfato de calcio hidratado, formado al reaccionar parte del SO4H2 con la lechada de cal 4 N añadida.
Las restantes causas de pequeña variación de volumen (como las del SO4H2 al pasar a SO4Na2 y SO4K2), se consideran sin sensible influencia.
Triturar con varilla de vidrio el sulfato de calcio precipitado y remover bien.
Diluir, si es necesario, la solución de cenizas hasta un contenido en bromo del vino de 1 mg/l, aproximadamente. Para comprobar cualitativamente si el contenido en bromo del vino es superior o inferior a 1 mg/1, tomar 1 ml de la solución de cenizas del vino, poner en tubo de ensayo y añadir una gota de solución tampón pH 4,65, una gota de solución rojo fenol y una gota de solución cloramina T. Después de un minuto exactamente, detener la reacción añadiendo una gota de solución de tiosulfato sódico. Si la solución es amarilla, amarillo-parda o amarillo-verdosa, el contenido en bromo es aproximadamente inferior a 1 mg/l, y si es azul, violeta o violeta fugaz, el contenido en bromo es superior a 1 mg/1, siendo necesario diluir la solución.
Decantar el líquido y trasvasar a un tubo de ensayo.
Poner en tubo de ensayo 5 ml de la solución de cenizas y añadir 0,25 ml de solución tampón, 0,25 ml de rojo fenol y 0,25 mililitros de cloramina T.
Después de un minuto exactamente, paralizar la reacción con 0,25 ml de tiosulfato. Determinar la absorbancia con fotocolorímetro o espectrofotómetro mediante medida a 584 nm.
Para obtener la curva patrón, tomar la solución de bromuro de potasio con 10 mg de bromo por litro, alícuotas de 0,25, 0,50, 0,75, 1,00, 1,25, 1,50, 2,00 y 2,50 ml y llevar a 5 ml con agua destilada.
El contenido en bromo de estas soluciones es, respectivamente, 0,5, 1,-1,5, 2,0, 2,5, 3, 4 y 5 mg/l.
Tratar cada una de estas soluciones de 5 ml como la solución de cenizas y hacer las correspondientes determinaciones en el espectrofotómetro a 584 nm.
32.5 Cálculo.
Calcular el contenido en bromo expresado en mg/l mediante comparación con la correspondiente curva patrón y teniendo en cuenta el factor de dilución.
32.6 Referencias.
1. Jaulmes, P., y Bruns, S. «Valoration du brome dans les vins». Annales des Falsifications et de Fraudes. 1952.
2. Saldoval, J. A., e Hidalgo, T. «Medida espectrofotométrica de reacciones coloreadas muy fugaces». Boletín I. N. I. A. 19, 60, 40.
33. BROMO ORGÁNICO
33.1 Principio.
El coeficiente de dilución del ácido monobromoacético entre el éter y el agua acidulada con ácido sulfúrico (que anula la ionización) es próximo a 5. Los ésteres del ácido monobromoacético son menos solubles en el agua que el mismo ácido, por lo que es más fácil su extracción por el éter. Tres lavados con un volumen de éter igual al del vino son suficientes para extraer todo el bromo orgánico.
33.2 Material y aparatos.
33.2 (1, 2, 3 y 4) como 32.2 (1, 2, 3 y 4).
33.2.5 Ampollas de decantación de 50 y 500 ml.
33.3 Reactivos.
33.3 (1, 2, 3, 4, 5, 6 y 7) como 32.3 (1, 2, 3, 4, 5, 6 y 7).
33.3.8 Ácido sulfúrico puro.
33.4 Procedimiento.
Colocar en una ampolla de decantación 20-200 l de vino (según la riqueza supuesta), añadir 5 g de SO4H2 puro por litro de vino, se añade después un volumen de éter igual al volumen de vino, agitar y decantar cuidadosamente el éter. Repetir tres veces esta operación. Filtrar el éter, evaporar en frío en cápsula de cuarzo que contenga 0,5 ml de NaOH al 50 por 100 y 1 ml de lechada de cal 4 N. Después de unas 24 horas de saponificación de los ésteres, durante las que permanece la cápsula tapada con un vidrio de reloj, incinerar progresivamente el residuo a la temperatura de 550. Arrastrar y disolver las cenizas como en 32, realizando también en igual forma la valoración colorimétrica del bromo.
33.5 Cálculo.
Calcular el contenido en bromo expresado en mg/l a partir de la correspondiente curva patrón.
34. ÁCIDOS MONOBROMOACÉTICO Y MONOCLOROACÉTICO DE ORIGEN ORGÁNICO
(Método cualitativo) (Provisional)
34.1 Principio.
Siendo fraudulenta la presencia de compuestos orgánicos monohalogenados, es suficiente para investigar el fraude un análisis cualitativo.
Extracción de compuestos monohalogenados en medio ácido con éter y transformación por adición de amonio en glicocola y sal de amonio por combinación del halógeno desplazado. Separación cromatográfica y revelado con NO3Ag, obteniéndose una mancha blanca para el cloruro de plata y otra negra para el bromuro de plata.
34.2 Material y aparatos.
34.2.1 Frascos de tapón esmerilado que ajuste perfectamente, de 50 ml de capacidad.
34.2.2 Ampolla de decantación de 250 ml.
34.2.3 Agitador mecánico de unas 150 agitaciones por minuto.
34.2.4 Papel Watman número 1 para cromatografía.
34.2.5 Pipetas, micropipetas de 0,2 ml capilares divididas en milésimas.
34.2.6 Secador de aire.
34.2.7 Campana o recipiente para cromatografía y accesorios.
34.2.8 Pulverizador todo de vidrio y pulverización fina.
34.2.9 Campana de luz infrarroja.
34.3 Reactivos.
34.3.1 Éter sulfúrico para análisis.
34.3.2 Solución de ácido sulfúrico: solución acuosa al 1/3 en volumen.
34.3.3 Amoníaco puro de densidad 0,910 (24° Be) sin cloruros.
34.3.4 Mezcla acetona-agua en la proporción 100/20 en volumen.
34.3.5 Revelador de nitrato de plata. Mezclar 85 mg de NO3Ag, 1 ml de agua, 5 ml de amoníaco concentrado y etanol hasta 200 ml.
34.3.6 Revelador de pirogalol. Disolver 13 mg de pirogalol en 200 ml de etanol. La solución puede durar tres días si se conserva fría y al abrigo de la luz.
34.3.7 Solución testigo de cloro. Disolver 100 mg de ClNH4 en agua y enrasar a 1.000 ml.
34.3.8 Solución testigo de bromo. Disolver 100 mg de BrNH1 en agua y enrasar a 1.000 ml.
34.4 Procedimiento.
34.4.1 Extracción del compuesto halogenado. Tomar 100 ml de vino, llevar el matraz de 500 ml con tapón esmerilado, añadir 5 ml de ácido sulfúrico (1/3) y, acto seguido, 100 ml de éter. Agitar brevemente hasta formación de ligera emulsión, destapar unos momentos el frasco y observar la facilidad de separación.
A veces, como especialmente sucede con vino de prensa, la separación es difícil, siendo necesario nueva adición de ácido sulfúrico y de éter, Observar si hay facilidad de separación y llevar el frasco a un agitador mecánico, agitando durante tres horas. Poner en ampolla de decantación de 250 ml de capacidad con el fin de separar el vino de la fracción etérea; eliminar el éter por completo filtrando rápidamente por ligera capa de algodón.
Añadir inmediatamente 10 ml de amoníaco, agitar fuertemente durante tres minutos, con el fin de iniciar la formación del aminoácido correspondiente (glicocola) y evitar la de otros compuestos. En este momento se forma la sal de amonio por combinación del halógeno desplazado.
Dejar reposar 24 horas para que la reacción sea completa y proceder a la cromatografía.
34.4.2 Análisis cromatográfico.‒Depositar con micropipeta sobre papel Whatman número 1 para cromatografía (28 × 15 centímetros) a lo largo de una línea paralela al borde inferior y situada a 2,5 cm de este borde, 0,02 ml de soluciones testigo de Cl y de Br, y de 0,02 a 0,06 ml, según el contenido en halogenado, de las muestras de vino, separando cada aplicación 2 centímetros por los menos. Enrollar el papel y colocar en el fondo de la cubeta con el disolvente y en la cámara o campana de cromatografía.
Retirar el papel de la cubeta cuando el frente del líquido ascendente se encuentre a 1 cm del borde superior del papel, y dejar secar. Pulverizar con la solución de nitrato de plata y, estando húmedo el papel, pulverizar nuevamente con solución de pirogalol. Para mayor uniformidad del revelado, conviene desecar el papel con lámpara infrarroja.
La mancha correspondiente al Cl es de color blanco y tiene un Rf de 0,29, y la mancha correspondiente al Br es de color marrón y tiene un Rf de 0,59.
34.5 Referencia.
1. Morales, J., y Vega, R. «Comunicación de los Métodos Internacionales de Análisis». O. I. V., número 245, 1967.
35. ÍNDICE DE PERMANGANATO
35.1 Principio.
Valoración con una solución de permaganato del poder reductor en frío de las sustancias polifenólicas del vino. Como todos los componentes fenólicos del vino, a igual concentración no reducen la misma cantidad de permanganato, se trata de una valoración global que da un índice, al que se denomina «índice de permanganato»: IMn.
Constituye una expresión del contenido en sustancias fenólicas que están en relación con los caracteres gustativos de dureza, astringencia o aterciopelado sin que la relación sea absoluta. Es una determinación especial para vinos tintos; en blancos es poco apreciable y se presta a confusiones por consumo de permanganato por otros componentes del vino, que aquí tiene una influencia relativa más marcada.
35.2 Material y aparatos.
Los elementos necesarios para una volumetría.
35.3 Reactivos.
35.3.1 Solución N/100 de MnO4K. Se conserva mal. Prepararla a partir de solución N/10.
35.3.2 Indicador de índigo. Preparar una solución de 3 g/1 de carmín índigo. Filtrar, tomar 50 ml de esa solución, añadir 50 ml de SO4H2 y agua hasta 1.000 ml. Mezclar bien.
35.4 Procedimiento.
Colocar en un vaso de 50 ml del indicador de índigo antes indicado y añadir 2 ml del vino. Inmediatamente adicionar gota a gota la solución de MnO4K N/100, y al final, cada vez más lentamente, hasta desaparición completa del color azul y aparición de amarillo oro.
En vinos nuevos, el viraje es menos neto, por lo que conviene tomar sólo 1 ml de muestra.
Hacer la prueba testigo para eliminar el permanganato por sustancias no polifenales, sustituyendo los 2 ml de vino por 2 ml de una solución de ácido tártrico de 5 g/l, neutralizada hasta la mitad con KOH y encabezada hasta 10° de alcohol.
35.5 Cálculo.
Calcular el índice de permanganato o el título de materias oxidables en frío del vino por permanganato expresado en meq/l.
Índice de permanganato = 5 (V ‒ V').
Materias oxidables en frío = 5 (V ‒ V') mea/l.
V = volumen en ml de solución MnO4K N/100 para oxidar el vino.
= volumen en ml de solución MnO4K N/100 gastados en la prueba testigo.
35.6 Referencia.
1. Ribereau-Gayon et Peynaud. «Analyse et Contróle des Vins», página 320, 1958.
36(a). DETERMINACIÓN DE PRESENCIA DE VINO PROCEDENTE DE «HÍBRIDOS PRODUCTORES DIRECTOS»
(Método cualitativo)
36(a).1 Principio.
Transformación de la malvina, por la acción controlada de un oxidante apropiado, en una sustancia que a la luz de Wood acusa viva fluorescencia de color verde muy sensible.
36(a).2 Material y aparatos.
36(a).2.1 Lámpara que emita luz fluorescente de 366 nm de longitud de onda (luz de Wood).
36(a).2.2 Tubos de ensayo.
36(a).3 Reactivos.
36(a).3.1 Ácido clorhídrico N.
36(a).3.2 Nitrito sódico al 1 por 100.
36(a).3.3 Alcohol con el 5 por 100 de NH3 (gas).
36(a).4 Procedimiento.
36(a).4.1 Vinos tintos.‒Para vinos tintos introducir en el tubo de ensayo: 1 ml de vino, una gota de C1H, N, y 1 ml de NO2Na al 1 por 100. Agitar y esperar dos minutos, después añadir 6 ml de alcohol con el 5 por 100 de NH3 (gas). Agitar, esperar cinco minutos, filtrar y observar el filtrado a la luz de Wood. Si hay malvina se apreciará la fluorescencia color verde.
36(a).4.2 Vinos rosados o claretes.‒Introducir en el tubo de ensayo 5 ml de vino, cinco gotas de ácido clorhídrico N y 1 ml de nitrito de sodio al 1 por 100.
Agitar, esperar dos minutos y añadir después alcohol con el 5 por 100 de amoníaco. Agitar y observar a la luz de Wood (366 nm).
36(a).5 Cálculo.
La intensidad de la fluorescencia verde es muy sensible y puede acusar netamente la presencia de una dosis de 0,2 por 100 de híbrido Oberlein en el vino, siendo la tolerancia admitida hasta un 2 por 100 de riqueza de híbrido Oberlein.
36(a).6 Referencia.
1 Ribereau-Gayon, J.; Peynaud, E. «Analyse et Contróle des Vins», pág. 320, 1958.
36(b). DETERMINACIÓN DE PRESENCIA DE VINO PROCEDENTE DE «HÍBRIDOS PRODUCTORES DIRECTOS»
(Provisional)
36(b).1 Principio.
Determinación de malvina en vinos tintos secos por cromatografía de papel. La fase móvil es la solución Britten-Robinson, en las condiciones del método. Observación del cromatograma a la luz de Wood de 366 nm.
Como escala de comparación se emplea una serie de manchas de soluciones de pinacianol, que da fluorescencia similar a la de la malvina, aunque con coloración verdosa en vez de la roja ladrillo de la malvina, pero con intensidades de la fluorescencia proporcionales a las dosis aplicadas, haciendo comparativos los resultados.
36(b).2 Material y aparatos.
36(b).2.1 Fuente de luz de Wood (366 nm).
36(b).2.2 Cámara o campana para cromatografía y accesorios (soporte, pinzas, etc.).
36(b).2.3 Papel Arches para cromatografía.
36(b).3 Reactivos.
36(b).3.1 Solución Britten-Robinson. Mezclar 3,92 g de ácido fosfórico puro, 2,40 g de ácido acético puro y 4,48 g de ácido bórico cristalizado puro y añadir agua destilada hasta 1.000 ml. El pH de esta solución es 1,81.
36(b).3.2 Pinacianol o azul de quinaldina (ioduro de 1-1 dietilcarbocianina).
36(b).3.3 Alcohol neutro de 96°. Destilar alcohol rectificado en presencia de sosa (5 g/l).
36(b).4 Procedimiento.
Hacer aplicaciones con micropipeta de 50 μl de vino cada una, sobre papel a 2 cm del borde inferior, de tal forma que aparezcan manchas rectangulares de 2 cm de base por 0,5 cm de los bordes. Aplicar 12 gotas de 4 μl en cada punto, secando cada vez con corriente de aire a temperatura no superior a 30°.
Llenar la cubeta hasta una altura de 2 cm con la solución Britten-Robinson. Sumergir el papel sólo 1 cm y retirarlo cuando el líquido llegue a 1 cm del borde superior. Ayudar a la desecación con corriente de aire no superior a 30° y terminar la desecación al ambiente.
Para obtener una escala patrón preparar una solución en alcohol neutro de 96° de pinacianol de 0,5 g/l y diluir a 1/10. Tomar alícuotas de 0,3, 0,4, 0,6, 0,8, 1,2, 1,7, 2,4, 3,5 y 5 ml y completar con alcohol puro hasta 5 ml. A estas mezclas les corresponde un contenido convencional de híbrido de 3, 4, 6, 8, 12, 17, 24, 35 y 50 por 100, respectivamente.
Aplicar cada una de estas mezclas como anteriormente. Secar las manchas, recortar y pegar mediante una cola celulósica (aplicar en pequeñas dosis para que no desborde) a lo largo del borde de una tira de cartulina negra.
Renovar la escala cada 30 ó 45 días, pues pierde intensidad, por lo que hay que conservarla al abrigo de la luz.
Observar las manchas del cromatograma a la luz de Wood (366 nm) en cámara oscura y hacer rápidamente la comparación con la escala.
Para comprobar la eficacia de la lámpara se hace el siguiente ensayo: Lavar por capilaridad con alcohol puro el papel Arches de cromatografía. Aplicar solución alcohólica de pinacianol de 1 mg/l en cantidad tal que la mancha sea visible y la misma cantidad diluida a la mitad de una mancha no visible en las mismas condiciones de observación. Regular la distancia de la lámpara a la hoja para conseguir este efecto.
36(b).5 Cálculo.
Si al observar el cromatograma del vino a la luz de Wood en las condiciones expuestas se aprecian manchas color rojo ladrillo y fluorescentes de un Rf = 0,55 ± 0,09 será clara manifestación de presencia de malvina.
Para el análisis cuantitativo, comparar con la escala patrón. La intensidad del color de cada mancha de la escala testigo de solución de pinacianol será valorada por el número de miligramos de pinacianol por litro de solución alcohólica con que se ha hecho la mancha sobre el papel.
Con esta definición convencional de la intensidad del color de las manchas puede establecerse también, por comparación de estas manchas con las correspondientes a las de cromatogramas de vinos con mezcla conocida de «híbrido», un coeficiente proporcional a la riqueza de éste presente en los mismos.
La sensibilidad de las manchas pueden acusar el 1 por 100 de híbrido Oberlein (elegido como patrón).
La dosis correspondiente al valor 2 de la escala convencional, o sea el 2 por 100 de híbrido Oberlein, es la máxima tolerada provisionalmente por la Convención Internacional.
36(b).6 Referencias.
1. Dorier y Verelle. «Comunication a la Société des Experts-Chemistes de France». 6 enero 1965.
2. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V. A18, 1-4, 1969.
37. METANOL
(Método del ácido cromotrópico)
37.1 Principio.
Oxidación del metanol a formol por permanganato potásico en presencia de ácido fosfórico y medida espectrofotométrica de la reacción coloreada del formol con ácido cromotrópico. Coloración violeta específica del formol.
37.2 Material y aparatos.
37.2.1 Matraces aforados de 50 ml.
37.2.2 Baño de agua con regulación a 60-75°.
37.2.3 Espectrofotómetro que permita lectura a 575 mμ.
37.2.4 Matraz esférico de cuello largo, de 500 ml y refrigerante con alargadera.
37.3 Reactivos.
37.3.1 Solución de permanganato potásico. Disolver 3 g de MnO4K y 15 ml de PO4H3 en 100 ml de agua. Preparar mensualmente.
37.3.2 Solución de sal sódica de ácido cromotrópico (1-8 dihidroxinaftaleno, 3-6 disulfónico). Solución al 5 por 100. Filtrar si no queda clara. Preparar semanalmente.
37.3.3 Solución al 2 por 100 P/V de sulfito neutro de sodio anhidro.
37.4 Procedimiento.
Diluir o ajustar la muestra hasta una concentración total de alcohol de 5-6 por 100 en volumen. Utilizando 50 ml de muestra, destilar en destilador simple, recogiendo 40 ml de destilado en baño de hielo. Diluir hasta 50 ml con agua (si se ha determinado previamente el alcohol, el destilado puede ajustarse a 5-8 por 100 de concentración de alcohol y utilizarse para esta prueba). Si hay más de 0,05 por 100 de metanol por volumen, diluir hasta aproximadamente esa concentración con alcohol del 5,5 por 100. Para muestras que contengan menos de 0,05 por 100 de metanol, poner 200 ml de destilador eficiente de fraccionamiento, colocar el sistema de destilación reflujo total durante 15 minutos y luego destilar lentamente a elevada proporción de reflujo (por lo menos 20 : 1). Recoger 10 ml de destilado y diluir hasta 160 ml con agua.
Pipetar 2 ml de solución de KMnO4 en un matraz aforado de 50 ml. Enfriar en baño de hielo, añadir.1 ml de muestra diluida y fría y dejar 30 minutos en baño de hielo. Decolorar con un poco de NaHSO5 seco y añadir 1 ml de solución de ácido cromotrópico. Añadir lentamente con agitado y en baño de hielo 15 ml de SO4H2 y colocar 15 minutos en baño de agua caliente (60-75°). Enfriar, añadir una cantidad suficiente de agua para llevar aproximadamente a la marca de 50 ml, mezclar y diluir hasta volumen con agua a temperatura ambiente. Leer la absorbancia a 575 nm utilizando como reactivo en blanco alcohol de 5,5 por 100 tratado análogamente a la forma descrita. Tratar solución patrón de metanol que contenga 0,025 por 100 por volumen de metano) en alcohol de 5,5 por 100 simultáneamente en la misma forma, y leer la absorbancia (la temperatura del patrón y de la muestra no debe diferir en más de ‒1°, ya que la temperatura afecta a la intensidad del color).
37.5 Cálculo.
Calcular el contenido en metanol expresado en porcentaje.

A = absorbancia de la muestra.
A = absorbancia de la solución patrón de metanol.
F = factor de dilución de la muestra.
37.6 Observaciones.
Si el color de la muestra es demasiado intenso, diluir con reactivo en blanco. No diluir más de tres veces porque la relación de ácido cromotrópico a metanol se reduce demasiado.
37.7 Referencia.
1. «Association of Official Agricultural Cremists» Official Méthods of Analysis, pág. 138, 1965.
38. NITRÓGENO
(Método Kjeldhal)
38.1 Principio.
Transformación del nitrógeno orgánico en sulfato de amonio por ataque con ácido sulfúrico en ebullición en presencia de sustancias catalizadoras que acortan el tiempo de la mineralización y adición de sal potásica que eleve el punto de ebullición. Destilación por ebullición de vapor recogiendo el destilado en volumen conocido de solución de ácido sulfúrico.
Valoración con solución de tiosulfato sódico, del iodo liberado por el ácido sulfúrico al añadir al destilado solución de ioduro-iodato potásico.
38.2 Material y aparatos.
38.2.1 Aparato micro-Kjeldahl.
38.3 Reactivos.
38.3.1 Ácido sulfúrico puro, d = 1,84.
38.3.2 Catalizador de selenio. Mezclar bien pulverizados 80 g de SO1K2, 20 g de SO4Cu y 5 g de selenio.
38.3.3 Solución de fenolftaleína y papel tornasol.
38.3.4 Ácido sulfúrico N/50.
38.3.5 loduro potásico, solución al 4 por 100.
38.3.6 Iodato potásico, solución al 5 por 100.
38.3.7 Solución de tiosulfato sódico N/50.
38.3.8 Engrudo de almidón.
38.4 Procedimiento.
Poner en el matraz Kjeldahl 50 ml de vino, evaporar a ebullición lenta hasta unos 10 ml, añadir 10 ml de SO4H2 puro y 0,6 gramos de la mezcla pulverizada y bien uniformada de SO4Cu, selenio y SO4K2, continuar calentando primero lentamente y después enérgicamente, hasta decoloración, y proseguir unos minutos más. Si es necesario se van añadiendo nuevas dosis de SO4H2 puro de 5 en 5 ml.
Enfriar, pasar el contenido a matraz aforado de 100 ml arrastrando el residuo con pequeñas porciones de agua destilada, para enjuagar varias veces el matraz, comprobando que los últimos lavados no dan trazas de reacción ácida al papel de tornasol.
Tomar una alícuota de 20 ml y llevar al matraz del aparato de destilación, adicionando unas gotas de fenolftaleína. En un vaso cilíndrico de unos 200 ml, se añaden 10 ml de SO4H2 N/50. Añadir al matraz solución de sosa al por 100 en cantidad suficiente para viraje de la fenolftaleína y se encaja rápidamente en su sitio. Conectar el aparato de destilación con el generador de vapor de agua y comenzará el arrastre del amoníaco pasando al refrigerante y recogiendo el destilado en la solución sulfúrica.
Después de 15-20 minutos, comprobar con papel tornasol si una gota del destilado no acusa reacción alcalina. En este caso separar el vaso y proceder a la valoración.
Añadir al vaso con el sulfúrico N/50 ml de ioduro de potasio al 4 por 100 y 2 ml de iodato de potasio al 5 por 100 Mezclar con una varilla y después de 20 minutos valorar el iodo liberado con solución de tiosulfato sódico N/50 empleando como indicador 5-10 ml de solución de almidón.
38.5 Cálculo.
Calcular el nitrógeno expresado en meq/l o en mg/I.


V = volumen en ml de solución de tiosulfato N/50.
39. FLÚOR
(Provisional)
39.1 Principio.
Incineración de 525º del vino en presencia de un exceso de calcio o magnesio, destilación en corriente de vapor del ácido fluorhídrico a 135-140°, valoración del ácido destilado a pH 3,6 por solución de nitrato de torio en presencia de alizarina sulfonato de sodio.
39.2 Material y aparatos.
39.2.1 Aparato de Truhaut, que consta de generador de vapor, barbotador de pared doble, ampolla para la introducción de la solución sulfúrica de las cenizas magnéticas, refrigerante de retorno y refrigerante de West para la salida del destilado (figura 39.I.).
39.2.2 Cápsula de platino o níquel para las cenizas.
39.2.3 Horno eléctrico regulable para 525°.
39.2.4 Matraz de 250 ml.
39.2.5 Dos cápsulas de porcelana iguales, de unos 10 cm de diámetro.
39.2.6 Material para volumetría.
39.3 Reactivos.
39.3.1 Solución tampón de pH = 3,1. Mezclar 10o ml de solución molar de acetato de sodio, 303 ml de agua destilada y 97 ml de ácido clorhídrico N/1.
39.3.2 Solución N/100 de nitrato de torio, Disolver 1,3806 g de nitrato de torio con 4 moléculas de agua, en un litro de agua.
39.3.3 Solución N/10 de hidróxido sódico.
39.3.4 Ácido sulfúrico al 1/10.
39.3.5 Indicador de azul de bromotimol.
39.3.6 Óxido de magnesio al 1/10.
39.3.7 Acetato de magnesio al 1/10.
39.3.8 Solución Spadns. Añadir a 0,02 g de Spadns, agua hasta 100 ml. El Spadns resulta de la combinación del ácido p-sulfamílico con la sal de sodio del ácido cromotrópico (ácido dihidroxi, 1,8, naftalenodisulfónico 3,6). Su proceso de preparación es laborioso. Se encuentra preparado por Easman-Kodak.
39.3.9 Sulfato de plata en cristales.
39.4 Procedimiento.
39.4.1 Incineración.‒Poner 20 ml de vino en cápsula de platino c de níquel, añadir 2 ml de suspensión de óxido de magnesio al 1/10 y 4 ml de acetato de magnesio al 1/10. Evaporar, desecar y calcinar en el horno a 550 °C.
39.4.2 Destilación.‒Colocar en el barbotador del aparato Truhaut 20 ml de ácido sulfúrico al 50 por 100, 0,5 g de lana o polvo de vidrio y 0,5 g de sulfato de plata, con el fin de retener el C1H. Llevar el barbotador a 146 °C por ebullición del tetracloroetano (punto de ebullición 146°) en la doble pared (A) y forzar la corriente de vapor de agua a la velocidad de 5 g por minuto. Conviene calentar previamente el tetracloroetano en la doble pared del aparato antes de que barbote el vapor en el líquido, por haberse observado que así se evitan la formación de burbujas de espuma, que son un peligro por posible desborde y paso al refrigerante de salida.
Se continúa la destilación hasta recoger unos 100 ml de destilado.
Con estas operaciones queda el aparato desprovisto de posible presencia de flúor.
Disolver las cenizas en el menor volumen posible de SO4H2 al 1/10 e introducir en el barbotador, enfriando previamente el líquido del barbotador antes de verter en él la solución sulfúrica de cenizas. Proseguir la destilación con el barboteo de vapor, comenzando por calentar previamente, como se ha indicado, el tetracloroetano contenido en la doble pared. Recoger unos 100 ml de destilado en un matraz de 250 ml que contenga 5 ml de agua, de forma que quede sumergido el pico de la alargadera del refrigerante.
39.4.3 Valoración.‒Descarbonatar el destilado por agitación en frío y con vacío neutralizar con Na (OH) N/10 en presencia de azul de bromotimol como indicador. Llevar después a una cápsula de porcelana blanca bien limpia, siendo recomendable frotar previamente su interior con pasta silícea y enjuagarla con agua destilada. Añadir a 50 ml del destilado 1 ml de solución acuosa de Spadns y 1 ml de solución tampón de pH = 3,1. Añadir con microbureta solución N/100 de nitrato de torio hasta viraje a rojo violeta.
Para apreciar el punto del viraje se compara la tonalidad del color con la de una valoración testigo hecha en otra cápsula igual y con el mismo volumen de solución tampón, 1 ml de Spadns y 0,05 ml de solución de nitrato de torio. Se ha observado que con 0,1 ml de nitrato de torio es más clara la comparación de colores.
Restar del volumen de nitrato de torio gastado en el líquido problema los 0,05 y 0,1 ml añadidos al testigo.
39.5 Cálculo.
Equivalencia: 1 ml de solución de nitrato de torio N/100 < > 0,19 mg de flúor.
Expresar los resultados en mg de flúor por litro y con una aproximación de unos 0,5 mg.
39.6 Referencias.
1. Fabré, R.; Trumant, A.; Bouquet, A., y Bernuchon, J. (1960-1965), Cont. Rendu Aca. Sciences. 1965, 240, pág. 211; 1959, 248, pág. 504.
El barbotador (C) tiene una pared doble (A) donde se hierve el tetracloroetano a 146°. El tubo de entrada de vapor de agua rodea (E) la pared interna y penetra en su interior, prolongándose verticalmente hasta casi el fondo.
El refrigerante de retorno para condensar los vapores de tetracloroetano se conecta en (B).
La ampolla (F), con llave y tapón esmerilado, se ajusta a la boca del barbotador y el tubo penetra hasta la misma altura que el punto de entrada del tubo de vapor de agua. La salida del destilado comunica con refrigerante de West, vertical que se prolonga por alargadera tubular cuyo pico se sumerge en el agua que contiene el matraz receptor.
40. HIDROXIMETILFURFURAL
(Provisional)
40.1 Principio.
El hidroximetilfurfural se produce al calentar en exceso los mostos para su concentración, desulfitación o pasterización. La presencia de hidroximetilfurfural en ciertos vinos dulces se considera como indicio de la adición de mostos.
Este método está basado en la reacción de Winckler (formación de un complejo rojo para la acción del hidroximetilfurfural sobre la p-toluidina y ácido barbitúrico). Se utiliza para los vinos blancos y tintos, previa extracción con éter sulfúrico en los segundos.
40.2 Material y aparatos.
40.2.1 Espectrofotómetro y accesorios (cubeta para 1 cm de espesor del líquido).
40.2.2 Matraces aforados.
40.2.3 Embudo de decantación con tapón esmerilado y de 100 ml de capacidad.
40.2.4 cápsula de porcelana de unos 10 cm de diámetro.
40.3 Reactivos.
40.3.1 Reactivo de p-toluidina. Disolver 10 g de p-toloudina purísima en 50 ml de isopropanol. Llevar a matraz aforado de 100 ml, añadir 10 ml de ácido acético y completar hasta el enrase con isopropanol. Este reactivo dura 4 días.
40.3.2 Reactivo de ácido barbitúrico. Se introduce en matraz aforado de 100 ml, 0,5 g de ácido barbitúrico desecado a 105 °C y unos 70 ml de agua. Se calienta ligeramente en baño de agua, se enfría y enrasa con agua.
40.4 Procedimiento.
Llevar 10 ml del vino a embudo de decantación y añadir 12 ml de éter sulfúrico. Agitar durante 3 minutos, decantar el éter a cápsula de porcelana que contenga 2 ml de agua destilada, repetir cuatro veces la extracción del residuo del embudo con otros 12 ml de éter cada vez y decantar igualmente a la cápsula. Dejar evaporar el éter en medio ambiente. Al residuo de evaporación añadir agua hasta 10 ml, agitar y tomar 2 ml para análisis.
Mezclar los 2 ml tomados con 5 ml de reactivo de p-toluidina y 1 ml de ácido barbitúrico, llevándolos inmediatamente a la cubeta del espectrofotómetro (de un cm de espesor del líquido) y hacer rápidamente la lectura de la absorbancia a 546 nm. Emplear como líquido de referencia el agua destilada en lugar de ácido barbitúrico, o sea, 3 ml de agua más 5 ml de reactivo de p-toluidina.
40.5 Cálculo.
Determinar el contenido en hidroximetilfurfural expresado en mg/l por comparación con la correspondiente curva patrón teniendo en cuenta el factor de dilución.
40.6 Observaciones.
En el caso de vinos turbios centrifugar previamente. Si hay presencia de CO2 eliminarlo agitando el vino en un matraz.
40.7 Referencias.
1. Sandoval, J. A.; Hidalgo, T. «Puesta a punto de un método para análisis cuantitativo espectrofotométrico de hidroximetilfurfural en los vinos». Boletín INIA, 16: 4. 454-469, 1967.
2. Amerine, M. A. «Hydroximethyl-furfural in California Wines». Food Research. 13: 264-269, 1948.
41. DETERMINACIÓN CUALITATIVA DE ÁCIDO SÓRBICO, BENZOICO p-CLOROBENZOICO, SALICÍLICO, p-HIDROXIBENZOICO Y SU ESTER ETÍLICO
41.1 Principio.
Extracción con éter de los antisépticos del vino previamente acidificados. Separación por cromatografía de capa fina. Identificación a la luz ultravioleta y reveladores específicos.
41.2 Material y aparatos.
41.2.1 Extractor en continua de Palkin (figura 41.1) que comporta un tubo A de 55 cm de longitud y 3,8 cm de diámetro. La parte superior va unida a un refrigerante de bolas mediante boca esmerilada 34-25. A 30 cm de la base y formando un ángulo de 60° se encuentra un tubo lateral acodado a los 13 cm, formando un ángulo de 120°. Este tubo lateral termina en una junta esmerilada 24/40 en bisel, lo que permite su unión a un matraz de 50 ml que se calienta sobre una placa. En el tubo A se introducen bolitas de vidrio y el tubo interior (B) de 48,5 cm de longitud y 1,2 cm de diámetro, terminando en una placa de vidrio poroso.
41.2.2 Agitador mecánico (ver 41.6.1).
41.2.3 Erlenmeyer de 500 ml de boca esmerilada.
41.2.4 Baño de María.
41.2.5 Estufa eléctrica (200 °C).
41.2.6 Embudos de separación de 500 ml.
41.2.7 Cápsulas de porcelana de 70-80 cm de diámetro.
41.2.8 Equipo de cromatografía de capa fina.
41.2.9 Placas de vidrio de 20 x 20 cm.
41.3 Reactivos.
41.3.1 Éter sulfúrico.
41.3.2 Metanol.
41.3.3 Alcohol de 96°.
41.3.4 Ácido sulfúrico diluido a 20 por 100 (v/v).
41.3.5 Sulfato de sodio anhidro.
41.3.6 Polvo de poliamida para cromatografía Macherey-Nagel o Merck.
41.3.7 Revelador F254 Merck o equivalente.
41.3.8 Solventes:
— Pentano n: 10 partes.
— Hexano n: 10 partes.
— Ácido acético glacial: 3 partes.
41.3.9 Soluciones patrones. Preparar soluciones de 0,1 g por 100 ml en alcohol de 96 °Con los ácidos sórbico, p-clorobenzoico, salicílico, p-hidroxibenzoico y sus ésteres. Preparar una solución de 0,2 g por 100 ml en alcohol de 96 °Con el ácido benzoico.
41.3.10 Preparación de las placas. 10 g de poliamida con el 3 por 100 de indicador fluorescente se homogeneizan durante 20 segundos en agitador mecánico con 35 ml de metanol y 25 ml de cloroformo. Las placas impregnadas se secan durante 1 hora a 80 °C.
41.3.11 Solución hidroalcohólica conteniendo 0,15 g por 100 de dicromato potásico (41.6.2).
41.3.12 Solución saturada de ácido tiobarbitúrico en etanol (0,1 por 10C aproximadamente) (41.6.2).
41.3.13 Solución de 0,5 g de p-nitroanilina en 100 ml de CH2N (41.6.3).
41.3.14 Solución de nitrito sódico al 5 por 100 (41.6.3).
41.3.15 Solución de acetato sódico al 20 por 100 (41.6.3).
41.3.16 Solución de carbonato sódico al 15 por 100 (41.6.3).
41.3.17 Reactivo de Pauly. Disolver 4,5 g de ácido sulfamílico en 45 ml de CIH (2 N, calentando, diluir la solución e 500 ml en agua, 16 ml de la solución diluida se enfrían con hielo), se le añaden 10 ml de solución acuosa de nitrito sódico 4,5 por 100 también fría. El reactivo resultante se mantiene durante 15 minutos a 0 °C (a esta temperatura es estable de 1 a 3 días) y se le añade un volumen igual de solución acuosa de carbonato sódico 10 por 100 en el momento de pulverizar las placas (41.6.4).
41.4 Procedimiento.
Acidular 100 ml de vino o mosto con 10 ml de ácido sulfúrico al 20 por 100.
Extraer tres veces con 20 ml de éter sulfúrico cada vez, reuniendo las soluciones etéreas; o bien, durante 6 horas en el extractor continuo de Palkin con 175 ml de éter a una velocidad de reflujo de 500-600 ml/hora.
Los extractos etéreos se desecan con 6 g de sulfato sódico anhidro durante 12 horas. Filtrar y el éter se evapora a sequedad en baño de María 40-45°. El residuo se disuelve en un ml de etanol absoluto tomando cantidades de 5 a 10 μl para cromatografía. Mediante el tratamiento de los extractos etéreos con 5 ml de NaOH 2,5 N y acidulado de nuevo con 5 ml de SO4H2 20 por 100 se obtendrá en los cromatogramas las manchas correspondientes a los ácidos liberados por efecto de la saponificación.
Para obtener el desarrollo de los cromatogramas, depositar la muestra en forma de manchas puntuales de 5-10 μl.
El desarrollo se lleva a cabo en cámara saturada con el eluyente y a temperatura ambiente. Dejar migrar el solvente hasta una altura de unos 15 cm (una hora y media a dos horas/día). Retirar la placa de la cubeta y dejar secar a la temperatura ordinaria.
41.5 Cálculos.
Observación a la luz ultravioleta (360 nm).
Los conservadores aparecen de abajo arriba en el siguiente orden: ácido p-hidroxibenzoico, ésteres del ácido p-hidroxibenzoico, ácido salicílico, ácido p-clorobenzoico, ácido benzoico y ácido sórbico.
Para detectar el ácido sórbico pulverizar primeramente el cromatograma con la solución 41.3.11 y seguidamente con la 41.3.12. Calentar la placa a 80 °C, 5-10 minutos. La mancha de ácido sórbico aparece de color rojo sobre fondo ocre.
Para detectar los ácidos salicílicos y p-hidroxibenzoico y éster etílico del ácido p-hidroxibenzoico, pulverizar la placa con una mezcla compuesta de 2 ml de 41.3.13 más 3-5 gotas de 41.3.14 más 8 ml de 41.3.15, añadiéndolos en este orden. Una vez seca la placa aparecen las manchas correspondientes a los tres componentes citados de color amarillo. La materia colorante propia de los vinos toma colores que van del amarillo al naranja. Pulverizar de nuevo con la solución 41.3.16. Las manchas del ácido p-hidroxibenzoico toman color rojo; las del éster etílico del ácido p-hi-droxibenzoico, color rosa, y las del ácido salicílico amarillo limón. Los distintos componentes de la materia colorante toman colores que van desde distintos tonos de ocre al pardo y también verde y azulado.
Para detectar los ácidos p-hidroxibenzoico y salicílico y el éster etílico del ácido p-hidroxibenzoico se utiliza empleando el reactivo 41.3.17.
41.6 Observaciones.
41.6.1 Para el análisis cualitativo.
41.6.2 Para detectar el ácido sórbico.
41.6.3 Para detectar los ácidos salicílico y p-hidroxibenzoico y éster etílico del ácido p-hidroxibenzoico.
41.6.4 Para detectar los ácidos p-hidroxibenzoico y salicílico y el éster etílico del ácido p-hidroxibenzoico.
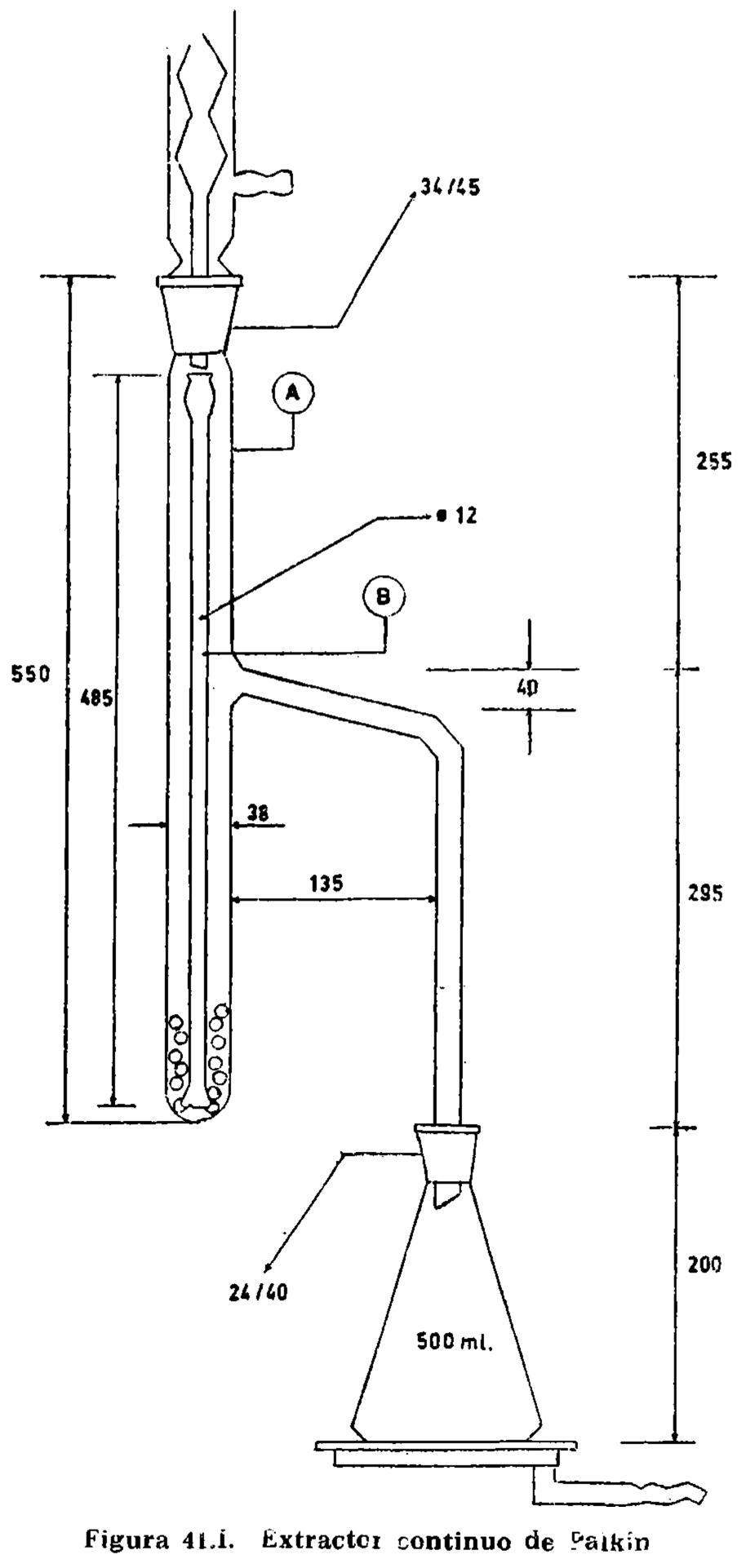
Vinagres
1. EXTRACTO TOTAL
1.1 Principio.
El extracto seco total se define como el conjunto de todas las sustancias que, en condiciones físicas determinadas, no se volatilizan. Estas condiciones físicas deben fijarse de tal manera que las sustancias componentes del extracto sufran el mínimo de alteración.
El método está basado en una evaporación del vinagre en baño de agua con secado posterior en estufa regulable a 100-105 grados centígrados.
1.2 Material y aparatos.
1 2.1 Balanza con aproximación de 0,001 g.
1 2.2 Estufa con ventilación de aire regulable de 100 a 105 °C.
1.2.3 Desecador de vidrio con ácido sulfúrico concentrado o gel de sílice (sílice amorfa impregnada con sustancia reveladora) como sustancia desecadora.
1.2.4 Baño de agua.
1.2.5 Cápsulas de fondo plano de 5 cm de diámetro construidas en platino, níquel o cuarzo.
1.2.6 Pipetas aforadas de 10 ml con doble enrase.
1.3 Procedimiento.
Agitar la muestra y filtrar a través de un papel de filtro plegado (tipo jarabe). Medir 10 ml del vinagre filtrado en una cápsula previamente tarada. Evaporar en baño de agua hirviente durante 30 minutos y mantener en la estufa durante dos horas y media a 105º C. Dejar secar la cápsula en el interior del desecador y, una vez fría, pesar en la balanza de precisión.
1.4 Cálculo.
El valor del extracto total del vinagre, en porcentaje, se hallará mediante la fórmula siguiente:
Extracto total: (M-m) 10
en la que:
M = masa, en gramos, de la cápsula con el extracto seco.
m = masa, en gramos, de la cápsula vacía.
1.5 Referencias.
1. Instituto de Racionalización del Trabajo. Una norma española 33.101,
2. A. O. A. C. Official Methods of Analysis. 11.ª ed., página 519, 30.081.
2. ACIDEZ TOTAL
(Grado acético)
2.1 Principio.
La acidez total se define como la totalidad de los ácidos volátiles y fijos que contienen el vinagre expresada en gramos de ácido acético por 100 ml de vinagre.
La determinación se efectúa mediante una volumetría de neutralización en presencia de solución alcohólica de fenolftaleína como indicador.
2.2 Material aparatos.
2.2.1 Bureta de 50 ml graduada en décimas de mililitro.
2.2.2 Pipeta aforada de 10 ml con doble enrase.
2.2.3 Matraces erlenmeyer de 250 ml de capacidad.
2.3 Reactivos.
2.3.1 Disolución de hidróxido sódico 0,5 N.
2.3.2 Solución indicadora de fenolftaleína. Se disuelven 10 g de fenolftaleína en alcohol etílico (93 a 96 por 100 en volumen) y se completa a un litro con el mismo solvente.
2.4 Procedimiento.
Medir 10 ml de vinagre, previamente filtrado, en un erlenmeyer de 250 ml. Diluir con 100 ó 150 ml de agua destilada recién hervida y fría hasta conseguir una solución débilmente coloreada. Añadir seis gotas de la solución indicadora de fenolftaleína y agregar solución de hidróxido sódico 0,5 N contenida en la bureta gota a gota y agitando hasta viraje del indicador.
2.5 Cálculos.
El valor de la acidez total o grado acético del vinagre en porcentaje se hallará mediante la fórmula siguiente:
Acidez total o grado acético = a.10.0,0300
en la que:
a = volumen, en ml, de solución NaOH 0,5 N.
2.6 Referencias.
1. Instituto de Racionalización del Trabajo. Una norma española 33.101.
2. A. O. A. C. Official Methods of Analysis. 11.ª ed., página 520, 30.068.
3. ACIDEZ FIJA
3.1 Principio.
La acidez fija se define como la totalidad de los ácidos fijos que contiene el vinagre, expresada en gramos, de ácido acético por 100 ml de vinagre.
3.2 Material y aparatos.
3.2.1 Baño de agua.
3.2.2 Bureta de 10 ml de capacidad graduada en décimas de mililitro.
3.2.3 Pipeta aforada de 10 ml con doble enrase.
3.2.4 Cápsulas de 200 ml de capacidad.
3.3 Reactivos.
3.3.1 Disolución de hidróxido sódico 0,1 N.
3.3.2 Disolución indicadora de fenolftaleína al 1 por 100 en alcohol.
3.4 Procedimiento.
Medir 10 ml de vinagre previamente filtrado y llevarlo a una cápsula de porcelana. Evaporar a sequedad en baño de agua. Añadir de 5 a 10 ml de agua destilada recientemente hervida y volver a evaporar a sequedad, repitiendo esta operación cinco veces más. Añadir, aproximadamente, 180 ml de agua destilada recientemente hervida y fría, añadir seis gotas de la solución indicadora de fenolftaleína y agregar solución de hidróxido sódico 0,1 N contenida en la bureta, gota a gota y agitando, hasta viraje del indicador.
3.5 Cálculos.
El valor de la acidez fija del vinagre, expresado en gramos de ácido acético por 100 ml, se hallará mediante la fórmula siguiente:
Acidez fija = a.10.0,00600
en la que:
a = volumen, en ml, de NaOH 0,1 N.
3.8 Referencias.
1. Instituto de Racionalización del Trabajo. Una norma española 33.101.
2. A. O. A. C. Official Methods of Analysis. 11.ª ed., página 520, 30.089.
4. ACIDEZ VOLÁTIL
4.1 Principio.
Se define convencionalmente como valor de la acidez volátil de un vinagre la diferencia entre los valores de su acidez total y fija, expresadas ambas en gramos de ácido acético por 100 ml de vinagre.
4.2 Cálculo.
El valor de la acidez volátil del vinagre, expresado en gramos de ácido acético por 100 ml, se hallará mediante la fórmula siguiente:
At ‒ Af = acidez volátil
en la que:
At = acidez total o grado acético, expresado en gramos de ácido acético por 100 ml.
Af = acidez fija de la misma muestra de vinagre, expresada en gramos de ácido acético por 100 ml.
4.3 Referencias.
1. Instituto de Racionalización del Trabajo. Una norma española 33.101.
2. A. O. A. C. Official Methods of Analysis. 11.ª ed., página 520, 30.070.
5. SULFATOS
5.1 Principio.
Precipitación del sulfato de bario en medio ácido.
5.2 Material y aparatos.
5.2.1 Vasos de precipitado de 250 ml.
5.2.2 Baño de agua.
5.2.3 Estufa.
5.2.4 Desecador.
5.3 Reactivos.
5.3.1 Acido clorhídrico N.
5.3.2 Cloruro bárico (1 g/100 ml de Cl2Ba 2H2O).
5.4 Procedimiento.
A 100 ml de muestra en vaso de precipitado de 250 ml añadir 2 ml de ClH normal, calentar a ebullición e ir añadiendo gota a gota 10 ml de cloruro bárico (1 g/100 ml de Cl2Ba 2H2O) sin que deje de hervir la solución. Continuar hirviendo cinco minutos manteniendo el volumen constante, añadiendo agua caliente siempre que sea necesario. Dejar en reposo hasta que el sobrenadante esté claro (una noche es suficiente, aunque no conviene sobrepasar ese tiempo). Filtrar por papel sin cenizas de poro fino, lavar con agua destilada caliente hasta que no dé reacción con nitrato de plata las aguas del lavado, secar, calcinar, enfriar en desecador y pesar.
5.5 Cálculo.
La proporción de sulfatos = 7,465 P g/l de SO4K2
siendo:
P = peso, en g, del producto de la calcinación.
5.6 Referencias
1. A. O. A. C. Official Methods of Analysis. 11.ª ed. (1970).
2. Instituto de Racionalización del Trabajo. Una norma española 33-104-73.
6. ACETILMETILCARBINOL
(Acetoina) (Provisional)
6.1 Principio.
Oxidación de acetilmetilcarbinol a diacetilo por el ion férrico. Formación de dimetilglioxima con hidroxilamina. Precipitación con sal de níquel.
6.2 Material y aparatos.
6.2.1 Aparato de destilación, según figura 6.1.
6.2.2 Baño de agua.
6.2.3 Placa filtrante (diámetro medio de los poros: de 20 a 40).
6.2.4 Estufa.
6.3 Reactivos.
6.3.1 Cloruro férrico al 30 por 100.
6.3.2 Trozo de plato poroso.
6.3.3 Solución de clorhidrato de hidroxilamina al 20 por 100.
6.3.4 Solución de acetato sódico al 20 por 100.
6.3.5 Solución de cloruro de níquel al 10 por 100.
6.3.6 Amoníaco de densidad (d = 0,910).
6.4 Procedimiento.
En un matraz de 250 ml se ponen 50 ml de vinagre, 50 solución de cloruro férrico al 30 por 100 y unos trocitos de plato poroso. La mezcla se destila en un aparato (según figura 6.1), recogiendo durante 50 ó 60 minutos de 60 a 70 ml de destilado en un matraz donde previamente se han puesto 2 ml de la solución de clorhidrato de hidroxilamina al 20 por 100, 3 cm3 de la solución de acetato sódico al 20 por 100, 1 ml de la solución de cloruro de níquel al 10 por 100 y amoníaco concentrado en exceso, según el grado acético del vinagre (aproximadamente 3 ml).
Calentar el destilado, que quedará ligeramente amoniacal, treinta minutos en baño de agua hirviendo, y después de doce horas recoger el precipitado rojo de dimetilglioxima niquelosa sobre una placa filtrante tarada previamente, lavar con agua caliente, secar a 100° en estufa durante una hora y pesar, repitiendo la operación hasta obtener peso constante.
6.5 Cálculo.
Acetilmetilcarbinol - 12,2 P g/l
siendo:
P = peso, en mg, del precipitado.
6.6 Referencia.
1. Bull Technique de la Vinaeigreire, núm. 6, pág. 164, 1950.
7. CENIZAS
7.1 Principio.
Se denominan cenizas de un vinagre el conjunto de los productos de incineración del residuo de evaporación de un volumen conocido del vinagre, realizada de manera que se puedan obtener todos los cationes en forma de carbonatos y otras sales minerales.
7.2 Materia y aparatos.
7.2.1 Horno eléctrico regulable.
7.2.2 Baño de agua y baño de arena.
7.2.3 Lámpara de infrarrojo.
7.2.4 Cápsula de platino o de cuarzo de 70 mm de diámetro y 25 mm de altura de fondo plano.
7.3 Procedimiento.
Colocar 20 ml de vinagre en una cápsula tarada en balanza que aprecie 1/10 de mg. Evaporar con precaución en baño de agua hasta consistencia siruposa, continuar el calentamiento sobre baño de arena con moderación y durante una media hora. Es conveniente ayudar a la evaporación con la aplicación de rayos infrarrojos hasta carbonización. Cuando ya no se desprendan vapores llevar la cápsula al horno eléctrico a 525º ± 25º C y con aireación continua.
Después de cinco minutos de carbonización completa, sacar la cápsula del horno, dejar enfriar y añadir 5 ml de agua, que se evaporan en baño de agua, y llevar de nuevo al horno a 525 grados centígrados.
Si la combustión de las partes carbonosas no se consigue en 15 minutos, volver a comenzar la operación de adición de agua, evaporación y recalcinación.
Después de enfriar en el desecador, pesar la cápsula con las cenizas.
7.4 Calculos.
Calcular el contenido en cenizas expresado en g/l.
Cenizas = 50 P g/l
P= peso, en g, de las cenizas contenidas en 20 ml de vinagre.
Dar los resultados con una aproximación de 0,03 g/l.
7.5 Referencias.
1. Recueil des Méthodes Internationales d'Analyse des Vins. O. I. V., AGI, 1969.
2. Métodos Oficiales de Análisis de Vinos, Ministerio de Agricultura, 1971, pág. 66.
8. ÍNDICE DE OXIDACIÓN
8.1 Principio.
Oxidación con permanganato potásico de las sustancias volátiles fácilmente oxidables de los vinagres.
8.2 Material y aparatos.
8.2.1 Matraz de destilación de 250 ml de capacidad provisto de dispositivo de entrada de vapor de agua para destilación por arrastre de vapor.
8.2.2 Matraz erlenmeyer de 50 ml de capacidad provisto de boca esmerilada y tapón.
8.2.3 Matraz aforado de 50 ml de capacidad.
8.2.4 Refrigerante de serpentín de 25 cm de capacidad útil.
8.2.5 Calderín generador de vapor.
8.2.6 Pipeta aforada con doble enrase de 10, 20, 25, y 50 ml de capacidad.
8.2.7 Bureta de 50 ml de capacidad graduada en decimas de mililitro.
8.3 Reactivos.
8.3.1 Disolución de permanganato potásico aproximadamente 1 N.
8.3.2 Disolución valorada de tiosulfato sódico 0,5 N.
8.3.3 Disolución de yoduro potásico al 50 por 100. Ver 8.6.2.
8.3.4 Disolución indicadora de almidón. Se hace una papilla con 5 g de almidón soluble y 20 ml de agua y se vierte en 1 ml de agua hirviente. Se continúa la ebullición durante cinco minutos y se deja enfriar antes de su empleo. Conviene guardarla en frascos esterilizados.
8.3.5 Disolución de ácido sulfúrico al 50 por 100 en volumen.
8.4 Procedimiento.
Ajustar el vinagre a 4 g por 100 de acidez total, expresada en ácido acético. Tomar 50 ml de vinagre, con la acidez debidamente ajustada e introducir en el matraz de destilación. Pasar a su través una corriente de vapor de agua procedente del calderín descrito, a una velocidad regulada, de manera que cuando se hayan recogido 50 ml de destilado se conserven en el matraz de destilación aproximadamente 45 ml. Comprobar que la temperatura del destilado no sobrepase 25º C. Pasar los 50 ml del destilado al matraz erlenmeyer de 500 ml. Añadir 10 ml de la solución de ácido sulfúrico y 25 ml de la disolución de permanganato potásico. Mantener el matraz durante una hora en el baño de agua a 25º C.
Transcurrido ese tiempo, añadir 20 ml de la disolución de yoduro potásico. Tapar el matraz y mezclar bien su contenido. Inmediatamente valorar el yodo liberado con la disolución de tiosulfato sódico. Simultáneamente, a la muestra de vinagre llevar, en las mismas condiciones, un blanco en que el destilado se sustituya por 50 ml de agua destilada.
8.5 Cálculos.
El valor del índice de oxidación se hallará mediante la fórmula siguiente:

donde:
I = índice de oxidación.
a = volumen, en ml, de tiosulfato sódico consumido en la valoración de la muestra en blanco.
b = volumen, en ml, de tiosulfato sódico consumido en la valoración de la muestra de vinagre diluido.
8.6 Observaciones.
8.6.1 Si el índice de oxidación resulta mayor que 15, ha de repetirse la determinación, usando 25 ml de vinagre ajustando a 4 grados acéticos, añadiendo 25 ml de agua. Repetir esta reducción a la mitad hasta que el índice de oxidación sea menor que 15. Calcular el índice de oxidación, teniendo en cuenta las diluciones efectuadas.
8.6.2 Esta disolución no se debe usar si presenta color amarillento.
8.7 Referencias.
1. Instituto de Racionalización del Trabajo. Una norma española 33-102-73.
2. Métodos Oficiales de Análisis de la Association of Official Agricultural Chemists de Estados Unidos (ed. 1970).
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid





