Agencia Estatal Boletín Oficial del Estado






Las Órdenes de 30 de noviembre de 1976 («Boletín Oficial del Estado» de 4 de enero de 1977), de 31 de julio de 1979 («Boletín Oficial del Estado» de 29 y 30 de agosto), de 17 de septiembre de 1981 («Boletín Oficial del Estado» de 14 de octubre), de 1 de diciembre de 1981 («Boletín Oficial del Estado» de 20 de enero de 1982) y 18 de julio de 1989 («Boletín Oficial del Estado» del 25) establecieron diversos métodos oficiales de análisis de fertilizantes.
Como consecuencia de la plena integración de España en la Comunidad Económica Europea se hace necesario armonizar la legislación nacional con la correspondiente comunitaria en lo referente a los métodos de toma de muestras y análisis de los abonos adaptándolos a lo dispuesto en la Directiva de la Comisión 89/519/CEE, de 1 de agosto de 1989 («Diario Oficial de las Comunidades Europeas» número 265, de 12 de septiembre de 1989).
Esta armonización sobre los métodos de toma de muestras y análisis de los abonos prevé que se tomen muestras de estos productos de conformidad con los métodos comunitarios a fin de comprobar las condiciones sobre la calidad y composición de los abonos, así como eliminar los obstáculos técnicos a los intercambios en el sector, de acuerdo todo ello con las normas comunitarias.
El contenido de la presente norma determina requisitos sanitarios, por lo que la misma se dicta, sin perjuicio de la existencia de otros títulos competenciales, en aplicación de lo dispuesto en el artículo 40.2 de la Ley 14/1986, de 25 de abril, General de Sanidad y al amparo del artículo 149.1.16.a de la Constitución Española.
En su virtud, a propuesta de los Ministros de Economía y Hacienda; de Industria, Comercio y Turismo; de Agricultura, Pesca y Alimentación, y de Sanidad y Consumo, previo informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria y oídos los representantes de las organizaciones afectadas, de acuerdo con el Consejo de Estado, previa deliberación del Consejo de Ministros en su reunión del día 19 de julio de 1991,
DISPONGO:
Se aprueban como oficiales los métodos de análisis de fertilizantes que se detallan en el anexo.
Cuando no existan métodos oficiales para determinados análisis de fertilizantes y hasta tanto los mismos no sean aprobados por el Órgano competente y previamente informados por la Comisión Interministerial para la Ordenación Alimentaria, podrán ser utilizados los aprobados por los Organismos nacionales o internacionales de reconocida solvencia.
Lo dispuesto en el presente Real Decreto y los métodos oficiales que aprueba se dictan de acuerdo con lo establecido en el artículo 40.2 de la Ley 14/1986, de 25 de abril, General de Sanidad, y al amparo del artículo 149.1.16.ª de la Constitución Española.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan al presente Real Decreto y en particular el método número 10(c) que figura en la Oden de 30 de noviembre de 1976 («Boletín Oficial del Estado» de 4 de enero de 1977), los métodos números 25, 26, 27, 31, 34(a) y 34(b) que figuran en la Orden de 31 de julio de 1979 («Boletín Oficial del Estado» de 29 y 30 de agosto) y los métodos números 24(a), 24(b), 24(c) y 24(d) de la Orden de 18 de julio de 1989 («Boletín Oficial del Estado» de 25 de julio de 1989).
Dado en Madrid a 22 de julio de 1991.
JUAN CARLOS R.
El Ministro de Relaciones con las Cortes y de la Secretaría del Gobierno,
VIRGILIO ZAPATERO GÓMEZ
24(a). Extracción del calcio total, del magnesio total, del sodio total y del azufre total en forma de sulfato.
24(b). Extracción del azufre total presente en diversas formas.
24(c). Extracción de las formas solubles en agua del calcio, del magnesio, del sodio y del azufre en forma de sulfato.
24(d). Extracción del azufre soluble en agua, presente en diveras formas.
24(e). Extracción y determinación cuantitativa del azufre elemental.
24(f). Determinación manganimétrica del calcio extraído por precipitación en forma de oxalato.
24(g). Determinación cuantitativa del magnesio por espectrometría de absorción atómica.
24(h). Determinación cuantitativa del magnesio por complexometría.
24(i). Determinación cuantitativa de los sulfatos.
24(j). Determinación cuantitativa del sodio extraído.
24(a). Extracción del calcio total, del magnesio total, del sodio total y
del azufre total en forma de sulfato
24(a).1 Principio.–Disolución en ácido clorhídrico diluido mediante ebullición del calcio total, del magnesio total, del sodio total y del azufre total en forma de sulfato, para efectuar, en la medida de lo posible, una única extracción a partir de la cual poder determinar cada uno de estos elementos.
Este método es aplicable a los abonos CEE en los que, según lo dispuesto por la Directiva 89/284/CEE, hay que declarar el calcio total, el magnesio total, el sodio total y el azufre total en forma de sulfato.
24(a).2 Material y aparatos.–Placa calefactora eléctrica de temperatura regulable.
24(a).3 Reactivos.–Ácido clorhídrico diluido (1:1).
Un volumen de ácido clorhídrico (densidad 1,18) por un volumen de agua destilada.
24(a).4 Procedimiento:
24(a).4.1 Preparación de la muestra. Ver método número 2.
24(a).4.2 Toma de muestra. El calcio, el magnesio, el sodio y el azufre de los sulfatos se determinarán a partir de la muestra de 5 g que se habrá pesado con precisión de 1 mg.
No obstante, cuando el contenido en azufre (S) del abono sea superior al 15 por 100, esto es, un 37,5 por 100 de SO3, y el contenido en calcio (Ca) sea superior al 18,8 por 100, esto es, un 26,3 por 100 de CaO, el calcio y el azufre se determinarán a partir de una muestra de 1 g que se habrá pesado con precisión de 1 mg.
Las muestras se introducirán en vasos de precipitado de 600 ml.
24(a).4.3 Disolución. Añadir aproximadamente 400 ml de agua y 50 ml de ácido clorhídrico diluido [24(a).3] en pequeñas porciones y con precaución si el producto contiene una cantidad importante de carbonatos. Hervir y mantener en ebullición durante treinta minutos. Dejar enfriar agitando de vez en cuando. Trasvasar cuantitativamente a un matraz aforado de 500 ml. Añadir agua hasta alcanzar este volumen. Homogeneizar por agitación. Filtrar a través de un filtro seco en un recipiente seco. Desechar las primeras porciones. El extracto ha de estar perfectamente límpido. Tapar si el filtrado no va a utilizarse inmediatamente.
24(a).5 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método 8.1.
24(b). Extracción del azufre total presente en diversas formas
24(b).1 Principio.–Extracción del azufre total presente en los abonos en forma elemental y/o en diversas formas, transformándolo en medio fuertemente alcalino en polisulfuros y tiosulfatos, y oxidación de éstos y de los sulfitos eventualmente presente con peróxido de hidrógeno. Las diversas formas de azufre se transforman así en sulfato que se cuantifica en forma de sulfato de bario precipitado [método 24(i)].
El presente método se aplica a los abonos CEE en los que, según lo dispuesto en la Directiva 89/284/CEE, hay que declarar el azufre total presente en diversas formas (elemental, tiosulfato, sulfito, sulfato).
24(b).2 Material y aparatos.–Placa calefactora eléctrica de temperatura regulable.
24(b).3 Reactivos:
24(b).3.1 Ácido clorhídrico diluido (1:1). Un volumen de ácido clorhídrico (densidad 1.18) por un volumen de agua destilada.
24(b).3.2 Solución de hidróxido de sodio al 30 por 100 de NaOH como mínimo (densidad 1,33).
24(b).3.3 Solución de peróxido de hidrógeno al 30 por 100 en masa.
24(b).3.4 Cloruro de bario BaCl22H2O, solución acuosa de 122 g/l.
24(b).4 Procedimiento:
24(b).4.1 Preparación de la muestra: Ver método número 2.
24(b).4.2 Toma de muestra: Pesar, con precisión de 1 mg, una cantidad de abono que contenga entre 80 y 350 mg de azufre (S), esto es, de 200 a 875 mg de SO3.
En la mayoría de los casos (S<15 por 100), se pesarán 2,5 g.
Introducir la muestra en un vaso de precipitado de 400 ml.
24(b).4.3 Oxidación. Añadir 20 ml de solución de hidróxido de sodio [24(b).3.2] y 20 ml de agua. Tapar con un vidrio de reloj. Hervir durante cinco minutos sobre la placa calefactora [24(b).2]. Retirar de la placa. Arrastrar el azufre adherido a las paredes del vaso de precipitado con un chorro de agua hirviendo. Hervir y mantener en ebullición durante veinte minutos. Dejar enfriar.
Añadir el peróxido de hidrógeno [24(b).3.3] en dosis de 2 ml hasta que desaparezca la reacción. Normalmente son necesarios de 6 a 8 ml de peróxido de hidrógeno. Dejar que prosiga la oxidación en frío durante una hora, luego hervir durante media hora. Dejar enfriar.
24(b).4.4 Preparación de la solución problema. Añadir aproximadamente 50 ml de agua y 50 ml de la solución de ácido clorhídrico [24(b).3. l ]:
Si el contenido de azufre (S) es inferior al 5 por 100: Filtrar la solución en un vaso de precipitado de 600 ml. Lavar repetidas veces con agua fría el residuo del filtro. Al terminar el lavado, comprobar que no queden sulfatos en las últimas gotas del filtrado con ayuda de la solución de cloruro de bario [24(b).3.4]. El filtrado debe estar perfectamente límpido. La determinación cuantitativa de los sulfatos se efectuará en la totalidad del filtrado siguiendo el método 24(i).
Si el contenido en azufre (S) es del 5 por 100 o más: Trasvasar cuantitativamente el contenido del vaso de precipitado a un matraz aforado de 250 ml. Enrasar con agua. Homogeneizar por agitación. Filtrar a través de un filtro seco en un recipiente seco. El filtrado debe estar perfectamente límpido. Tapar el recipiente si la solución no va a utilizarse inmediatamente. La determinación cuantitativa de los sulfatos se efectuará en una alícuota de esta solución por precipitación en forma de sulfato de bario siguiendo el método 24(i).
24(b).5 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método 8.2.
24(c). Extracción de las formas solubles en agua del calcio, del
magnesio, del sodio y del azufre presente en forma de sulfato
24(c).1 Principio.–Disolución, en agua hirviendo, del calcio, del magnesio, del sodio y del azufre en forma de sulfato, para efectuar, en la medida de lo posible, una única extracción a partir de la cual poder determinar la cantidad de cada uno de estos elementos contenida en los abonos.
Este método es aplicable a los abonos CEE en los que, según lo dispuesto por la Directiva 89/284/CEE, hay que declarar el calcio, el magnesio, el sodio y el azufre en forma de sulfato, solubles en agua.
24(c).2 Material y aparatos.–Placa calefactora eléctrica de temperatura regulable.
24(c).3 Reactivos.–Agua destilada o desmineralizada de calidad equivalente.
24(c).4 Procedimiento:
24(c).4.1 Preparación de la muestra. Ver método número 2.
24(c).4.2 Toma de muestra:
24(c).4.2.1 Abono sin azufre o que contenga simultáneamente, y a lo sumo, un 3 por 100 de azufre (S) (= 7,5 por 100 SO3) y un 4 por 100 de calcio (Ca) (= 5,6 por 100 CaO): Pesar, con precisión de 1 mg, 5 g de abono.
24(c).4.2.2 Abono que contenga más de un 3 por 100 de azufre (S) y de un 4 por 100 de calcio (Ca): Pesar, con precisión de 1 mg, 1 g de abono.
Introducir en un vaso de precipitado de 600 ml la muestra pesada según [24(c).4.2.1] o [24(c).4.2.2].
24(c).4.3 Disolución. Añadir aproximadamente 400 ml de agua. Hervir durante treinta minutos. Dejar enfriar agitando de vez en cuando. Trasvasar cuantitativamente a un matraz aforado de 500 ml. Enrasar con agua. Homogeneizar por agitación. Filtrar, a través de un filtro seco, en un recipiente seco. Desechar las primeras porciones. El filtrado ha de estar perfectamente límpido.
Tapar el recipiente si la solución no va a utilizarse inmediatamente.
24(c).5 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 26 5, de 12 de septiembre de 1989. Método 8.3.
24(d). Extracción del azufre soluble en agua, presente en diversas formas
24(4).1 Principio.–Disolución en agua fría del azufre presente en diversas formas (tiosulfato, sulfito y sulfato) y transformación en sulfato por oxidación con peróxido de hidrógeno en medio alcalino.
Este método es aplicable a los abonos en los que, según lo dispuesto en la Directiva 89/284/CEE, hay que declarar el azufre soluble en agua presente en diversas formas (tiosulfato, sulfito, sulfato).
24(d).2 Material y aparatos:
24(d).2.1 Matraz aforado de 500 ml.
24(d).2.2 Agitador rotativo regulado 34/40 rpm.
24(d).2.3 Placa calefactora eléctrica de temperatura regulable.
24(d).3 Reactivos:
24(d).3.1 Ácido clorhídrico diluido (1:1). Un volumen de ácido clorhídrico (densidad 1,18) por un volumen de agua destilada.
24(d).3.2 Solución de hidróxido sódico al 30 por 100 de NaOH como mínimo (densidad 1,33).
24(d).3.3 Solución de peróxido de hidrógeno al 30 por 100 en masa.
24(d).4 Procedimiento:
24(d).4.1 Preparación de la muestra. Ver método número 2.
24(d).4.2 Toma de muestra:
24(d).4.2.1 Abono que contenga simultáneamente y a lo sumo, un 3 por 100 de azufre (S) (= 7,5 por 100 SO3) y un 4 por 100 de calcio (Ca) (= 5,6 por 100 de CaO): Pesar 5 g de abono con precisión de 1 mg.
24(d).4.2.2 Abono que contenga más de un 3 por 10 de azufre (S) y de un 4 por 100 de calcio (Ca): Pesar 1 g de abono con precisión de 1 mg.
Introducir en un matraz aforado de 500 ml [24(d).2.1] la muestra pesada según [24(d).4.2.1] o [24(d).4.2.2].
24(d).4.3 Disolución. Añadir aproximadamente 400 ml de agua.
Tapar. Agitar [24(d).2.2] durante treinta minutos. Enrasar con agua. Homogeneizar por agitación.
Filtrar a través de un filtro seco en un recipiente seco. Tapar si la solución no va a utilizarse inmediatamente.
24(d).4.4 Oxidación de la alícuota que va a analizarse. Tomar una alícuota de la solución de extracción que no sea superior a 50 ml y que contenga entre 20 y 100 mg de azufre (S). Introducirla en un vaso de precipitado de capacidad adecuada. En caso necesario llevar el volumen a unos 50 ml con agua.
Añadir 3 ml de solución de hidróxido de sodio [24(4).3.2] y 2 ml de solución de peróxido de hidrógeno [24(d).3.3].
Tapar con un vidrio de reloj y dejar hervir moderadamente durante una hora sobre la placa calefactora [24(d).2.3]. Añadir, en dosis de 1 ml. solución de peróxido de hidrógeno, hasta que cese la reacción (a lo sumo 5 ml).
Dejar enfriar, retirar y lavar el vidrio de reloj del vaso de precipitado. Acidificar con aproximadamente 20 ml de ácido clorhídrico diluido [24(d).3.1]. Añadir agua hasta completar 300 ml. Efectuar la determinación cuantitativa de los sulfatos de toda la solución oxidada siguiendo el método 24(i).
24(d).5 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método 8.4.
24(e). Extracción y determinación cuantitativa del azufre elemental
24(e).1 Principio.–Extracción del azufre elemental con sulfuro de carbono, previa eliminación de los compuestos solubles. Gravimetría del azufre extraído.
Este método es aplicable a los abonos CEE en los que según lo dispuesto por la Directiva 89/284/CEE, hay que declarar el azufre total en forma elemental.
24(e).2 Material y aparatos:
24(e).2.1 Matraz de extracción de 100 ml de cuello esmerilado.
24(e).2.2 Aparato de Soxhlet, con sus correspondientes cartuchos filtrantes.
24(e).2.3 Evaporador rotativo del vacío.
24(e).2.4 Estufa eléctrica con ventilación, regulada a 90 ± 2 ºC.
24(e).2.5 Cápsulas de Petri de porcelana, con un diámetro de 5 a 7 cm y una altura no superior a 5 cm.
24(e).2.6 Placa calefactora eléctrica de temperatura regulable.
24(e).3 Reactivos:
24(e).3.1 Sulfuro de carbono.
24(e).4 Procedimiento:
24(e).4.1 Preparación de la muestra. Ver método número 2.
24(e).4.2 Extracción del azufre. Introducir de 5 a 10 g de muestra, pesados con precisión de 1 mg, en un cartucho del aparato de Soxhlet [24(e).2.2].
Lavar a fondo, con agua caliente, el contenido del cartucho, para eliminar todos los compuestos solubles. Secar en estufa a 90 ºC [24(e).2.4] durante, al menos, una hora.
Introducir el cartucho en el aparato Soxhlet [24(e).2.2]. Después de introducir en el matraz [24(e).2.1] unas cuantas perlas de vidrio, se tara (Po) y se introducen 50 ml de sulfuro de carbono [24(e).3.1].
Conectar el aparato y extraer el azufre elemental durante seis horas.
Suprimir el calentamiento, dejar enfriar y desconectar el matraz del aparato. Poner el matraz en el evaporador rotativo [24(e).2.3] hasta que el contenido del matraz se solidifique en una masa esponjosa.
Secar el matraz en la estufa a 90 °C [24(e).2.4] hasta obtener un peso constante (P1). Suele bastar con una hora.
24(e).4.3 Determinación de la pureza del azufre extraído. Es posible que se extraigan otras sustancias, a la vez que el azufre elemental, con el sulfuro de carbono. Para determinar su proporción, se procederá de la siguiente forma:
Después de homogeneizar lo mejor posible el contenido del matraz, tomar de 2 a 3 g de sustancia y pesar con una precisión de 1 mg (n). Introducirlos en una cápsula de Petri [24(e).2.5] y pesarlo todo (P2). Ponerlo luego sobre la placa calefactora [24(e).2.6] regulada de tal forma que no supere los 220 ºC para no provocar una combustión del azufre. Continuar la sublimación entre tres y siete horas hasta alcanzar un peso constante (P3).
Nota: Para algunos abonos no hará falta conocer el estado de pureza del azufre, en cuyo caso el procedimiento concluirá al final del punto [24(e).4.2].
24(e).5 Cálculos.–El porcentaje de azufre elemental (S0) del abono es igual a:

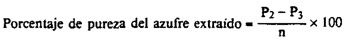
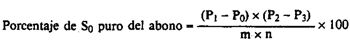
siendo:
m = la masa, en g, de la muestra de abono.
P0 = la masa, en g, del matraz de Soxhlet.
P1 = la masa, en g, del matraz de Soxhlet y del azufre impuro, una vez seco.
n = la masa, en g, del azufre impuro utilizado para la purificación.
P2 = la masa, en g, de la cápsula de Petri + la toma de muestra utilizada en el punto [24(e).4.3].
P3 = la masa, en g, de la cápsula de Petri después de la sublimación del azufre.
24(e).6. Observaciones.–En este método de análisis se utiliza sulfuro de carbono (CS2). Ello obliga a tomar disposiciones de seguridad especiales, en particular:
Almacenamiento de CS2.
Equipo de protección del personal.
Higiene en el trabajo.
Protección contra incendios y explosiones.
Eliminación del reactivo.
Para llevar a cabo este método se requiere un personal altamente cualificado y el equipo de laboratorio adecuado.
24(e).7 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método número 8.5.
24(f). Determinación manganimétrica del calcio extraído por precipitación en forma de oxalato
24(f). 1 Principio.–Precipitación del calcio presente en una alícuota de la solución de extracción en forma de oxalato. Una vez separado y disuelto, valorar el ácido oxálico con permanganato potásico.
Este método es aplicable a los abonos en los que según lo dispuesto por la Directiva 89/284/CEE, hay que declarar el calcio total y/o soluble en agua.
24(f).2. Material y aparatos:
24(f).2.1 Crisol filtrante de vidrio sinterizado con una porosidad de 5 a 20 µ.
24(f).2.2 Baño María.
24(f).3 Reactivos:
24(f).3.1 Ácido clorhídrico diluido (I:1). Un volumen de ácido clorhídrico (densidad 1,18) por un volumen de agua destilada.
24(f).3.2 Ácido sulfúrico diluido (I:10). Un volumen de ácido sulfúrico (densidad 1,84) por 10 volúmenes de agua destilada.
24(f).3.3 Solución de amoniaco (I:1). Un volumen de amoniaco (densidad 0.88) por un volumen de agua destilada.
24(f).3.4 Solución saturada de oxalato de amonio [(NH4)2.C2O4 (H2O)], a temperatura ambiente (40 g/l aproximadamente).
24(f).3.5 Solución de ácido cítrico al 30 por 100 (m/v).
24(f).3.6 Solución de cloruro de amonio al 5 por 100 (m/v).
24(f).3.7 Solución de azul de bromotimol al 0,1 por 100 (m/v) en etanol de 95 por 100.
24(f).3.8 Solución de verde de bromocresol al 0,04 por 100 (m/v) en etanol de 95 por 100.
24(f).3.9 Solución valorada de permanganato potásico 0,02 M.
24(f).4 Procedimiento:
24(f).4.1 Preparación de la alícuota que va a analizarse. Con una pipeta de precisión, tomar una alícuota de la solución de extracción obtenida por el método [24(a) o 24(c)], que contenga de 15 a 50 mg de Ca (= 21 a 70 mg de CaO). Siendo V2 el volumen de esta alícuota. Introducirla en un vaso de precipitado de 400 mI. Añadir unas gotas de solución del indicador [24(f).3.7]. Neutralizar, si fuera necesario, con unas gotas de la solución de amoniaco [24(f)3.3] (viraje del indicador de amarillo o azul).
Añadir 1 ml de la solución de ácido cítrico [24(f).3.5] y 5 ml de la solución de cloruro de amonio (24(f).3.6].
24(f).4.2 Precipitación del oxalato cálcico. Añadir aproximadamente 100 mI de agua. Hervir, añadir de 8 a 10 gotas de solución del indicador [4(f).3.8] y, gota a gota, 50 ml de solución caliente de oxalato de amonio [24(f).3.4]. Si se forma un precipitado, disolverlo añadiendo unas gotas de ácido clorhídrico [24(f).3.1]. Neutralizar muy lentamente con la solución de amoniaco [24(f).3.3], agitando continuamente hasta alcanzar un pH de 4,4 a 4,6 (viraje del indicador [24(f).3.8] de verde a azul. Poner el vaso de precipitado al baño María [24(f).2.2] y mantenerlo hirviendo durante treinta minutos. Sacar el vaso de precipitado del baño María, dejarlo en reposo durante una hora y filtrar sobre el crisol [24(f).2.1].
24(f).4.3 Valoración del oxalato precipitado. Lavar el vaso de precipitado y el crisol hasta eliminar por completo el exceso de oxalato de amonio (lo que se podrá verificar comprobando la ausencia de cloruro en el agua de lavado). introducir el crisol en el vaso de precipitado de 400 ml y disolver el precipitado con 50 mI de ácido sulfúrico caliente (24(f).3.2]. Añadir agua al vaso de precipitado hasta 100 ml aproximadamente.
Calentar hasta alcanzar una temperatura de 70 a 80 ºC y valorar, gota a gota, con la solución de permanganato [24(f).3.9] hasta que el color rosa se mantenga durante un minuto.
Siendo n este volumen.
24(f).5 Cálculos.–El contenido en calcio (Ca) del abono es igual a
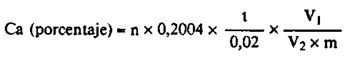
siendo:
n = los ml de permanganato utilizados.
m = la masa, en g, de la muestra.
V2 = el volumen, en ml, de la alícuota.
V1 = el volumen, en ml, de la solución de extracción.
t = la molaridad de la solución de permanganato en moles por litro.
CaO (porcentaje) = Ca (porcentaje) x 1,400
24(f).6 Referencias.–Directiva de la Comisión 89/519/CEE. «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método número 8.6.
24(g). Determinación cuantitativa del magnesio por espectrometría de absorción atómica
24(g).1 Principio.–Una vez diluido el extracto de forma adecuada, se determina el magnesio por espectrometria de absorción atómica.
Este método es aplicable a los abonos CEE, en los que, según lo dispuesto por la Directiva 89/284/CEE, hay que declarar el magnesio total y/o el magnesio soluble en agua, exceptuando los abonos del anexo de la Directiva 89/284/CEE sobre elementos secundarios: Tipo 4 (kieserita), tipo 5 (sulfato de magnesio) y tipo 7 (kieserita con sulfato de potasio), a los que se aplicará el método [24(h)].
El presente método se aplica a todos los extractos de abonos que contengan elementos en cantidad tal que puedan interferir en la determinación complexométrica del magnesio.
24(g).2 Material y aparatos:
24(g).2.1 Espectrómetro de absorción atómica provisto de una lámpara de magnesio y regulado a 285,2 nm. Llama de aire/acetileno.
24(g).3 Reactivos:
24(g).3.1 Ácido clorhídrico diluido aproximadamente 1M.
24(g).3.2 Ácido clorhídrico diluido aproximadamente 0,5 M.
24(g).3.3 Solución patrón de magnesio de 1,00 mg por ml.
24(g).3.3.1 Disolver 1,013 g de sulfato de magnesio (Mg.SO4.7H2O) en la solución de ácido clorhídrica 0,5 M [24(g).3.2] y seguir añadiendo este ácido hasta alcanzar 100 ml.
24(g).3.3.2 Alternativa 1: Pesar 1,658 g de óxido de magnesio (MgO) previamente calcinado para eliminar las trazas de recarbonatación. Introducirlo en un vaso de precipitado con 100 ml de agua y 120 ml de ácido clorhídrico 1 M [24(g).3.1]. Una vez disuelto, trasvasar cuantitativamente a un matraz aforado de 1.000 ml, enrasar con agua y homogeneizar por agitación.
24(g).3.3.3 Alternativa 2: Solución patrón comercial.
El laboratorio es responsable del control de estas soluciones comerciales.
24(g).3.4 Solución de cloruro de estroncio. Disolver 75 g de cloruro de estroncio (SrCl2.6H2O) en una solución de ácido clorhídrico diluido 0.5 M [24(g).3.2] y llevar a 500 ml con este mismo ácido.
24(g).4 Procedimiento:
24(g).4.1 Preparación de la solución problema [ver métodos 24(a) y 24(c)].
24(g).4.2 Si el contenido declarado en magnesio (Mg) del abono es superior al 6 por 100 (=10 par 100 de MgO), tomar 25 ml (V1) de la solución de extracción [24(g),4.1] e introducirla en un matraz aforado de 100 ml.
Enrasar con agua. Homogeneizar. El factor de dilución será D1=100/V1.
24(g).4.3 Con una pipeta, tomar 10 ml de la solución de extracción [24(g).4.1] o de la solución [24(g).4.2] e introducirla en un matraz aforado de 200 ml. Enrasar con la solución de ácido clorhídrico 0,5 M [24(g).3.2]. Homogeneizar. Factor de dilución 200/10.
24(g).4.4 Diluir la solución [24(g).4.3] con la solución de ácido clorhídrico 0,5 M [24(g).3.2]. hasta obtener una concentración correspondiente al rango óptimo de medidas del espectrómetro [24(g).2.1]. Siendo V2 el volumen de la extracción en 100 ml. El factor de dilución será D2= 100/V2.
La solución final deberá contener un 10 por 100 V/V de la solución de cloruro de estroncio [24(g).3.4].
24(g).4.5 Preparación de la solución en blanco.
Preparar una solución en blanco siguiendo todo el procedimiento desde la extracción [métodos de extracción 24(a) o 24(c)], omitiendo únicamente la toma de muestra del abono.
24(g).4.6 Preparación de las soluciones para trazar la curva patrón.
Diluir la solución patrón [24(g).3.3] con ácido clorhídrico 0,5 M, [24(g).3.2], preparar como mínimo cinco soluciones patrón de concentración creciente que correspondan al rango óptimo de medidas del aparato [24(g).2].
Estas soluciones deberán contener un 10 por 100 V/V de la solución de cloruro de estroncio [24(g),3.4].
24(g).4.7 Mediciones.
Preparar el espectrómetro [24(g).2.1] para medir a 285,2 nm.
Pulverizar, sucesivamente, las soluciones patrón [24(g).4.6], la solución problema [24(g).4.4] y la solución en blanco [24(g).4.5], lavando el aparato con la solución que vaya a medirse a continuación. Repetir esta operación tres veces.
Representar la curva patrón poniendo en ordenadas el valor de cada una de las soluciones patrón [24(g).4.6] en el espectrómetro y en abcisas las concentraciones en magnesio correspondientes expresadas en µg/ml. Partiendo de esta curva, determinar la concentración en magnesio de la muestra [24(g).4.4], esto es Xs, y la concentración de la solución en blanco [24(g).4.5], esto es Xb.
24(g).5 Cálculos.–Calcular la cantidad de magnesio (Mg) o de óxido de magnesio (MgO) de la muestra partiendo de las soluciones patrón y teniendo en cuenta el ensayo en blanco. El contenido en magnesio (Mg) del abono, en porcentaje, es igual a:
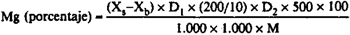
Xs = La concentración, en µg/ml, de la solución problema dada en la curva patrón.
Xb = La concentración, en µg/ml, de la solución en blanco dada en la curva patrón.
D1 = El factor de dilución después de la dilución especificada en el punto [24(g).4.2]. Este será igual a 4 cuando se extraigan 25 ml, y a 1 cuando no se haga dilución.
D2 = El factor de dilución del punto [24(g).4.4].
M = La masa, en g, de la muestra en el momento de la extracción.
MgO (porcentaje) = Mg (porcentaje)/0,6
24(g).6 Referencias.–Directiva de la Comisión 89/284/CEE, «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método número 8.7.
24(h). Determinación cuantitativa del magnesio por complexometría
24(h).1 Principios.–Disolución del magnesio contenido en los abonos por uno de los métodos 24(a) y/o 24(c). Primera valoración con EDTA de Ca+Mg en presencia de negro de eriocromo T. Segunda valoración con EDTA de Ca, en presencia de calceína o de ácido calconcarbónico. Determinación del Mg por diferencia.
El presente método es aplicable a los abonos CEE siguientes, para los que se establece la determinación cuantitativa del magnesio total y/o del magnesio soluble en agua:
Abonos de la Directiva 76/116/CEE: Abonos simples nitrogenados del tipo 1 b (nitrato de calcio y de magnesio), tipo 7 (sulfonitrato de magnesio), tipo 8 (abono nitrogenado con magnesio) y abonos simples potásicos del tipo 2 (sal bruta de potasa enriquecida), tipo 4 (cloruro de potasio con magnesio) y tipo 6 (sulfato de potasio con sal de magnesio).
Abonos que figuran en el anexo de la Directiva 89/284/CEE sobre elementos secundarios.
24(h).2 Material y aparatos:
24(h).2.1 Agitador magnético o mecánico.
24(h).2.2 Medidor de pH (pHmetro).
24(h).3 Reactivos:
24(h).3.1 Solución patrón de magnesio 0,05 M.
24(h).3.1.1 Disolver 1,232 g de sulfato de magnesio (MgSO4.7H2O) en la solución de ácido clorhídrico 0,5 M [24(h).3.11] y llevar a 100 ml con este mismo ácido.
O:
24(h).3.1.2 Pesar 2,016 g de óxido de magnesio previamente calcinado para eliminar cualquier recarbonatación. Introducirlos en un vaso de precipitado con 100 ml de agua. Añadir, mientras se agita, unos 120 ml de ácido clorhídrico aproximadamente 1 M [24(h).3.12]. Conseguida la disolución, trasvasar cuantitativamente a un matraz aforado de 1.000 ml, enrasar y homogeneizar. Un ml de dichas soluciones deberá contener 1,216 mg de Mg (=2,016 mg de MgO),
El laboratorio ha de controlar la concentración de esta solución patrón.
24(h).3.2 Solución 0,05 M de EDTA. Pesar 18,61 g de sal disódica dihidratada de ácido etilendiaminotetraacético (C10H14N2Na2Og.2H2O), introducirlos en un vaso de precipitado de 1.000 ml y disolver en unos 600 a 800 ml de agua. Trasvasar la solución cuantitativamente a un matraz aforado de 1.000 ml. Enrasar y homogeneizar. Controlar esta solución con la solución [24(h).3.1], extrayendo 20 ml de esta última y valorando según la técnica analítica descrita en el punto [24(h).5.3].
Un ml de solución de EDTA deberá corresponder a 1,216 mg de Mg (=2,016 mg de MgO) y a 2,004 mg de Ca (=2,804 mg de CaO) [ver las observaciones de los puntos 24(h).7.1 y 24(h).7.6].
24(h).3.3 Solución patrón de calcio 0,05 M. Pesar 5,004 g de carbonato de calcio, para análisis, en seco e introducirlos en un vaso de precipitado con 100 ml de agua. Añadir progresivamente, mientras se agita, 120 ml de ácido clorhídrico aproximadamente molar [24(h).3.2]. Hervir para expulsar el anhídrido carbónica, enfriar y trasvasar cuantitativamente a un matraz aforado de 1 I. Completar el volumen con agua y homogeneizar. Controlar la correspondencia de esta solución con la solución [24(h).3.2] según la técnica analítica [24(h).5.4].
Un ml esta solución deberá contener 2,004 mg de Ca (= 2,804 mg de CaO) y corresponder a 1 ml de la solución de EDTA 0,05 M (24(h).3.2].
24(h).3.4 Indicador calceína. Mezclar cuidadosamente en un mortero 1 g de calceína con 100 g de cloruro de sodio. Utilizar 0,010 g de esta mezcla. El indicador cambiará de verde a naranja. Se deberá valorar hasta que se obtenga un naranja sin reflejos verdes.
24(h).3.5 Indicador ácido calconcarbónico. Disolver 0,40 g de ácido calconcarbónico en 100 ml de metanol. Utilizar tres gotas de dicha solución. Esta solución se conserva estable durante cuatro semanas aproximadamente. El indicador cambiará de rojo a azul. Se deberá valorar hasta obtener un azul sin reflejos rojos.
24(h].3.6 Indicador negro de eriocromo T. Disolver 0,30 g de negro de eriocromo T en una mezcla de 25 ml de propanol-1 y de 15 ml de trietanolamina. Esta solución se conserva estable durante cuatro semanas aproximadamente. Utilizar tres gotas de esta solución. Este indicador cambiará de rojo a azul y deberá valorarse hasta que se obtenga un azul sin reflejos rojos. Sólo cambiará en presencia de magnesio. Si fuera necesario, añadir 1 ml de la solución patrón [24(h).3.1].
En presencia simultánea de calcio y magnesio, el EDTA se combinará primero con el calcio y, a continuación, con el magnesio. En tal caso, se valorarán conjuntamente ambos elementos.
24(h).3.7 Solución de cianuro potásico. Solución acuosa de KCN al 2 por 100 (no se puede pipetear con la boca). Véase [24(h).7.7].
24(h).3.8 Solución de hidróxido de potasio y de cianuro de potasio. Disolver 280 g de KOH y 66 g de KCN en agua, completar el volumen hasta un litro y homogeneizar.
24(h).3.9 Solución tampón pH 10,5. En un matraz aforado de 500 ml. disolver 33 g de cloruro de amonio en 200 ml de agua, añadir 250 ml de amoniaco (densidad = 0,91), enrasar con agua y homogeneizar, Comprobar con regularidad el pH de esta solución.
24(h).3.10 Ácido clorhídrico diluido 1:1. Un volumen de ácido clorhídrico (densidad 1,18) por un volumen de agua destilada.
24(h).3.11 Solución de ácido clorhídrico, aproximadamente 0,5 M.
24(h).3.12 Solución de ácido clorhídrico, aproximadamente 1 M.
24(h).3.13 Solución de hidróxido de sodio 5 M.
24(h).4 Prueba de control.–Efectuar una determinación en alicuotas de las soluciones [24(h),3.1] y [24(h).3.3], de tal forma que se tenga una relación Ca/Mg aproximadamente igual a la de la solución problema. A tal efecto, extraer (a) de la solución patrón [24(h).3.3] y (b-a) de la solución patrón [24(h).3.1], siendo (a) y (b) los ml de solución EDTA utilizados en las dos valoraciones efectuadas con la solución problema. Este procedimiento sólo es correcto si las soluciones de EDTA, de calcio y de magnesio son exactamente equivalentes. En caso contrario, deberán efectuarse las correcciones pertinentes.
24(h).5 Procedimiento:
24(h).5.1 Preparación de la solución problema. Ver métodos 24(a) y 24(c).
24(h).5.2 Alícuotas que han de extraerse. Dentro de lo posible, la alícuota deberá contener entre 9 y 18 mg de magnesio (=15 y 30 mg de MgO).
24(h).5.3 Valoración en presencia de negro de eriocromo T. Extraer con la pipeta una alícuota [24(h).5.2] de la solución problema e introducirla en un vaso de precipitado de 400 ml. Neutralizar, utilizando el pHmetro, el exceso de ácido con la solución de hidróxido de sodio 5 M [24(h).3.13]. Diluir con agua hasta unos 100 ml. Añadir 5 ml de solución tampón [24(h).3,9]. El pH medio con el pHmetro deberá ser de 10,5±0,1. Añadir 2 ml de solución de cianuro de potasio [24(h).3,7] y tres gotas de indicador negro de eriocromo T [24(h).3.6]. Valorar con la solución de EDTA [24(h).3.2], mientras se agita con moderación en el agitador [24(h).2.1] {ver los puntos [24(h).7.2], [24(h).7.3] y [24(h).7.4]}. Siendo «b» el número de ml de solución EDTA 0,05 M.
24(h).5.4 Valoración en presencia de calceína o de ácido calconcarbónico. Extraer con la pipeta una alícuota de la solución problema, igual a la utilizada para la valoración anterior, e introducirla en un vaso de precipitado de 400 ml. Neutralizar, utilizando el pHmetro, el exceso de ácido con la solución de hidróxido de sodio 5 M [24(h).3.13]. Diluir con agua hasta unas 100 ml. Añadir 10 ml de la solución de KOH-KCN [24(h).3.8] y el indicador [24(h).3.4] o [24(h).3.5]. Valorar con la solución de EDTA [24(h).3.2], mientras se agita con moderación en el agitador [24(h).2.1] {ver los puntos [24(h).7.2], [24(h).7.3] y [24(h).7.4]}. Siendo «a» el número de ml de solución EDTA 0,05 M.
24(h).6 Cálculos.–Para los abonos CEE a los que se aplica el presente método (5 g de abono por 500 ml de extracto), el contenido del abono es igual a:

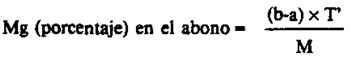
siendo:
a = Los ml de EDTA-0,05 M utilizados para la valoración en presencia de calceína o de ácido calconcarbónico.
b = Los ml de EDTA-0,05 M utilizados para la valoración en presencia de negro de eriocromo T.
M = La masa de la muestra, expresada en gramos, presente en la alícuota extraída.
T = 0,2016 x molaridad de la solución de EDTA/0,05 [ver 24(h).3.2].
T' = 0,1216 x molaridad de la solución de EDTA/0,05 [ver 24(h).3.2].
24(h).7 Observaciones:
24(h).7.1 La relación estequiométrica EDTA/metal en los análisis por coplexometría siempre es de 1:1, sea cual sea la valencia del metal y aunque el EDTA sea tetravalente. Por tanto, la solución de valoración de EDTA y las soluciones patrón, serán molares y no normales.
24(h).7.2 Los indicadores complexométricos suelen ser sensibles al aire. La solución puede perder color durante la valoración. Deberá añadirse entonces una o dos gotas de indicador. Esto ocurre sobre todo con el negro eriocromo T y el ácido calconcarbónico.
24(h).7.3 Los complejos metal-indicador son a veces relativamente estables y pueden tardar en virar. Por ello se deberán añadir lentamente las últimas glotas de EDTA y comprobar que no se ha superado el viraje añadiendo una gota de la solución 0,05 M de magnesio [24(h).3.1] o de calcio [24(h).3.3], especialmente para el complejo eriocromo-magnesio.
24(h).7.4 El viraje del indicador no deberá observarse de arriba abajo, sino horizontalmente a través de la solución, y el vaso de precipitado deberá colocarse sobre un fondo blanco en una posición adecuada con relación a la luz. El viraje también puede observarse fácilmente colocando el vaso de precipitado sobre un vidrio esmerilado, alumbrado por debajo con una luz moderada (lámpara de 25 W).
24(h).7.5 Este análisis requiere cierta experiencia y conviene ejercitarse, por ejemplo, observando los virajes de las soluciones patrón [24(h).3.1 y 24(h).3.3]. Es preferible que sea el mismo analista del laboratorio quien efectúe las determinaciones.
24(h).7.6 Para facilitar el control de la equivalencia entre las soluciones patrón [24(h).3.1, 24(h).3.2 y 24(h).3.3] se puede utilizar una solución de EDTA de concentración garantizada (tales como Trítisol o Normex).
24(h).7.7 No deben verterse a la red de alcantarillado las soluciones que contengan cianuro de potasio sin haber transformado previamente el cianuro en un compuesto inocuo, por ejemplo, por oxidación con hipoclorito sódico, después de alcalinizarlo.
24(h).8 Referencias.–Directiva de la Comisión 89/284/CEE, «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método 8.8.
24(i). Determinación cuantitativa de los sulfatos
24(1).1 Principio.–Determinación del azufre en forma de sulfato presente en extratos de abonos mediante determinación gravimétrica de los sulfatos por precipitación en forma de sulfato de bario.
Este método se aplica a la determinación cuantitativa de los sulfatos presentes en las extracciones efectuadas según los métodos 24(a), 24(b). 24(c) y 24(d).
24(i).2 Material y aparatos:
24(i).2.1 Cápsulas de porcelana para incineración.
24(i).2.2 Baño maría.
24(i).2.3 Estufa de secado regulada a 105 ºC ± 1 ºC.
24(i).2.4 Horno eléctrico, con circulación de aire, regulado a 800 ºC ± 50 ºC.
24(i).3 Reactivos:
24(i).3.1 Ácido clorhídrico diluido (1:1). Un volumen de ácido clorhídrico (densidad 1,18) por un volumen de agua destilada.
24(i).3.2 Cloruro de bario BaCl2.2H2O, solución acuosa de 122 g/l.
24(i).3.3 Nitrato de plata, solución acuosa de 5 g/l.
24(i).4 Procedimiento:
24(i).4.1 Preparación de la solución. Con una pipeta, tomar una alícuota de una de las soluciones de extracción mencionadas en el punto [24(i).1] que contenga entre 20 y 100 mg de S, es decir, entre 50 y 250 mg de SO3.
Introducir esta alícuota en un vaso de precipitado de capacidad adecuada. Añadir 20 ml de ácido clorhídrico diluido [24(i).3.1]. Añadir agua hasta 300 ml.
24(i).4.2 Obtención del precipitado. Hervir la solución, añadiendo, gota a gota, aproximadamente 20 ml de la solución de cloruro de bario [24(i).3.2], agitando enérgicamente la solución en el vaso de precipitado. Continuar la ebullición durante unos minutos.
Poner al baño maría hirviendo [24(i).2.2] el vaso de precipitado cubierto con un vidrio de reloj durante una hora. A continuación, dejar en reposo y en caliente (aproximadamente 60 ºC), hasta que el líquido sobrenadante quede claro. Retirar del baño maría y dejar enfriar.
Decantar la solución clara a través de un filtro sin cenizas, de filtración lenta. Lavar el precipitado varias veces por decantación con un volumen adecuado de agua caliente. Seguir lavando el precipitado en el filtro hasta la eliminación de los cloruros, lo cual se comprobará con la solución de nitrato de plata [24(i).3.3].
24(i).4.3 Calcinación y pesada del precipitado. Introducir el filtro con el precipitado en una cápsula de pocelana [24(i).2.1] previamente tarada con precisión de 0,1 mg. Secar en la estufa [24(i).2.3] y calcinar a 800 ºC [24(i).2.4] aproximadamente, durante media hora. Dejar enfriar en un desecador y pesar con precisión de 0,1 mg.
24(i).5 Cálculos.–Un mg de sulfato de bario corresponde a 0,137 mg de S o a 0,343 mg de SO3.
El contenido del abono en porcentaje de S es igual a:
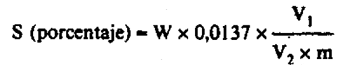
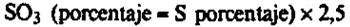
siendo:
W = El peso, en mg, del precipitado de sulfato de bario.
V1 = El volumen, en ml, de la solución de extracción.
V2 = El volumen, en ml, de la alícuota.
m = La masa, en g, de la muestra.
24(i).6 Referencias.–Directiva de la Comisión 89/519/CEE, «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método número 8.9.
24(j). Determinación cuantitativa del sodio extraído
24(j).1 Principio.–Tras haber diluido convenientemente el extracto obtenido con los métodos 24(a) y/o 24(c), se determinará por espectrometría de emisión de llama el contenido en sodio de la solución.
El presente método es aplicable a los abonos CEE en los que, según lo dispuesto en la Directiva 89/284/CEE, hay que declarar el sodio.
24(j).2 Material y aparatos:
24(j).2.1 Espectrómetro de emisión de llama regulado a 589,3 nm.
24(j).3 Reactivos:
24(j).3.1 Ácido clorhídrico diluido (1:1). Un volumen de ácido clorhídrico (densidad: 1,18) par un volumen de agua destilada.
24(j).3.2 Nitrato de aluminio, Al(NO3)3.9H2O.
24(j).3.3 Cloruro de cesio. CsCl.
24(j).3.4 Cloruro de sodio anhidro. NaCI.
24(j).3.5 Solución de cloruro de cesto y de nitrato de aluminio En un matraz aforado de 1.000 ml, disolver en agua 50 g de cloruro de cesio [24(j).3.3] y 250 g de nitrato de aluminio [24(j).3.2]. Enrasar con agua y homogeneizar por agitación.
24(j).3.6 Solución patrón de sodio de 1 mg/ml de Na. En un matraz aforado de 1.000 ml, disolver en agua 2,542 g de cloruro de sodio [24(j).3.4]. Añadir 10 ml de ácido clorhídrico [24(j).3.1]. Enrasar con agua. Homogeneizar por agitación.
24(j).3.7 Solución patrón de sodio de 40 µg/ml de Na. Introducir 10 ml de solución patrón [24(j).3.6] en un matraz aforado de 250 ml. Enrasar con agua. Homogeneizar por agitación.
24(j).4 Procedimiento.
24(j).4.1 Soluciones patrón. En matraces aforados de 100 ml introducir 0, 5, 10, 15, 20 y 25 ml de la solución intermedia [24(j).3.7]. Añadir 10 ml de la solución [24(j).3.5]. Enrasar con agua. Homogeneizar por agitación.
Concentración de las soluciones: 0, 2, 4, 6, 8 y 10 µg/ml de Na.
24(j).4.2 Preparación de las soluciones problema. Dependiendo del contenido en sodio previsible de la solución de extracción (5 g de abono en 500 ml) obtenida siguiendo el método 24(a) o el método 24(c); efectuar las diluciones según el siguiente cuadro:
|
(Porcentaje) Na2O |
(Porcentaje) Na |
Dilución intermedia |
Dilución final |
Factor dilución |
||
|---|---|---|---|---|---|---|
|
Alícuota (ml)(V2) |
Dilución en Ml (V3) |
Alícuota (ml)(V4) |
Dilución en ml |
|||
|
3- 5 |
2,2- 3,7 |
10 |
50 |
10 |
100 |
50 |
|
5-10 |
3,7- 7,4 |
10 |
100 |
10 |
100 |
100 |
|
10-20 |
7,4-15,0 |
10 |
100 |
5 |
100 |
200 |
|
20-38 |
15,0-28,0 |
5 |
100 |
5 |
100 |
400 |
La dilución intermedia se hará con agua. Para la dilución final se añadirán 10 ml de la solución [24(j).3.5] en el matraz aforado de 100 ml.
Para una muestra de 1 g, multiplíquese por 5 la alícuota de la dilución final (V4).
24(j).4.3 Mediciones. Preparar el espectrómetro [24(j).2.1] para efectuar mediciones de 589,3 nm. Calibrar el aparato midiendo la respuesta de las soluciones patrón [24(j).4.1]. Regular después la sensibilidad del aparato de forma que se utilice totalmente su escala cuando se emplee la solución patrón más concentrada. A continuación medir la respuesta de la solución de la muestra que vaya a analizarse [24(j).4.2]. Repetir la operación tres veces.
24(j).5 Cálculos.–Representar la curva patrón poniendo en ordenadas las medias de las respuestas de cada una de las soluciones patrón y en abcisas las concentraciones correspondientes expresadas en µg/ml. A partir de ésta, calcular la concentración en sodio de la solución de la muestra.
Calcular la cantidad de sodio a partir de las soluciones patrón teniendo en cuenta las diluciones. Expresar los resultados en porcentaje de la muestra.
El porcentaje en sodio (Na) del abono es igual a:
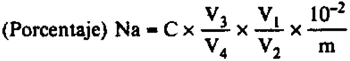
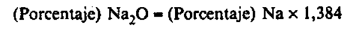
siendo:
C = La concentración, en µg/ml, de la solución introducida en espectrómetro.
V1 = El volumen, en ml, de la solución de extracción.
V2 = El volumen, en ml, de la alícuota en la disolución intermedia.
V3 = El volumen, en ml, de la dilución intermedia.
V4 = El volumen, en ml, de la alícuota en la dilución final (en 100 ml).
m = La masa, en g, de la muestra.
24(j).6 Referencias.–Directiva de la Comisión 89/519/CEE, «Diario Oficial de las Comunidades Europeas» número L 265, de 12 de septiembre de 1989. Método 8.10.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




