Agencia Estatal Boletín Oficial del Estado






El Real Decreto 72/1988, de 5 de febrero («Boletín Oficial del Estado» del 6), sobre fertilizantes y afines, en la redacción que al mismo ha dado el Real Decreto 877/1991, de 31 de mayo («Boletín Oficial del Estado» de 12 de junio), que lo modifica, define lo que se entiende por fertilizantes o abonos orgánicos y otras categorías afines destinadas directa o indirectamente a ser utilizadas en la agricultura y que por su contenido en alimentos nutritivos o por su acción específica tienden a facilitar, directa o indirectamente, el crecimiento de las plantas cultivadas, así como la producción de otros efectos que en aquel precepto se detallan. El párrafo segundo del artículo 5.º de este Real Decreto establece que los métodos de tomas de muestras y de análisis serán los oficiales.
Las Órdenes de 17 de septiembre de 1981 («Boletín Oficial del Estado» de 14 de octubre) y de 1 de diciembre de 1981 («Boletín Oficial del Estado» de 20 de enero de 1982), establecieron diversos métodos oficiales de análisis de productos orgánicos fertilizantes, siendo necesario complementarlos con otros nuevos que atiendan al control de diversos parámetros contemplados en el Real Decreto 72/1988, anteriormente aludido.
En su virtud, a propuesta de los Ministros de Economía y Hacienda; de Industria, Comercio y Turismo; de Agricultura, Pesca y Alimentación, y de Sanidad y Consumo, oídos los sectores afectados, previo informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros, en su reunión del día 12 de julio de 1991,
DISPONGO:
Se aprueban e incorporan como oficiales los métodos de análisis de productos orgánicos fertilizantes que se detallan en el anexo. Tales métodos se utilizarán para el análisis de todos los productos fertilizantes y afines de naturaleza total o parcialmente orgánica.
Cuando no existan métodos oficiales para determinados análisis, y hasta que no fueran aprobados por el Órgano competente y previamente informados por la Comisión Interministerial para la Ordenación Alimentaria, podrán ser utilizados los aprobados por los Organismos nacionales o internacionales de reconocida solvencia.
Dado en Madrid a 12 de julio de 1991.
JUAN CARLOS R.
El Ministro de Relaciones con las Cortes y de la Secretaría del Gobierno,
VIRGILIO ZAPATERO GÓMEZ
ÍNDICE
4. Extracto húmico total y ácidos húmicos.
7. Grado de finura.
8. Nitrógeno total.
9. Nitrógeno amídico (ureico).
10. Nitrógeno nítrico (método de Robertson).
12. Nitrógeno orgánico.
14. Fósforo soluble en agua y en citrato amónico (asimilable).
15. Nitrógeno insoluble en agua.
17. Potasio total.
18. Aminoácidos libres y totales.
4. EXTRACTO HÚMICO TOTAL Y ÁCIDOS HÚMICOS
En este método, las muestras se someten a una extracción alcalina para obtener el extracto húmico total y posteriormente se precipitan en este extracto los ácidos húmicos a pH: 1.
Tanto en el extracto húmico, como en el precipitado, se determina el contenido de carbono orgánico.
4.2.1 Tubos de centrífuga de 200 ml.
4.2.2 Baño de agua.
4.2.3 pH-metro.
4.2.4 Agitador de vaivén.
4.2.5 Centrífuga.
4.2.6 Estufa de aire forzado o normal.
4.3.1 Dicromato potásico 1N.
4.3.2 Ácido fosfórico 85 por 100.
4.3.3 Ortofenantrolina (0,5 g. se disuelven en 20 cc. de H2O y se añaden 100 cc. de SO4H2.
4.3.4 Sal de Mohr 0,5N.
4.3.5 Solución extractante de pirofosfato 0,1M –sosa 0,1N, recientemente preparada. Mezclar 44,5 g. de P2O7Na4 . 10H2O y 4 g. de NaOH, enrasando a un litro con agua.
4.3.6 Ácido sulfúrico (1:1).
4.3.7 Hidróxido sódico 0,5N.
4.3.8 Ácido sulfúrico 0,001N.
4.4.1 Extracción:
Desecar la muestra a 50-60 ° C curante cuarenta y ocho horas de estufa de aire forzado o a 100 ° C durante veinticuatro horas en estufa normal, pulverizando a continuación hasta obtener una muestra fina y homogénea.
Pesar, con precisión de 0,1 mg., la cantidad de muestra necesaria (ver 4.6.1) e introducirla en un tubo de centrífuga de 200 ml. de capacidad. Añadir 100 ml. de solución extractante 4.3.5 recientemente preparada. Agitar durante una hora, en agitador de vaivén, a la máxima velocidad posible y centrifugar a 4.500 r.p.m. durante veinticinco minutos como máximo. Repetir esta operación hasta que el líquido de extracción sea incoloro o ligeramente colorado (el número de repeticiones no debe pasar de cinco. Ver 4.6.2.)
Reunir todos los líquidos de las centrifugaciones en un matraz de un litro, enrasando con agua destilada. A esta solución se le denomina «Extracto húmico total».
4.4.2 Valoración del carbono orgánico del extracto húmico:
Tomar 50 ml. del extracto y llevar a un vaso o matraz de 500 ml., evaporando el baño de agua hasta sequedad. Añadir 10 ml. de solución normal de CR2O7K2 (4.3.1) y a continuación 20 ml. de ácido sulfúrico concentrado, agitando suavemente durante un minuto. Dejar reposar durante treinta minutos (tiempo de oxidación).
Transcurrido este tiempo, añadir 200 ml. de agua destilada y 10 ml. de PO4H3 (4.3.2), dejando enfriar. A continuación, añadir un ml. de difenilamina (4.3.3) y valorar con sal de Mohr (4.3.4), comparando con una prueba en blanco.
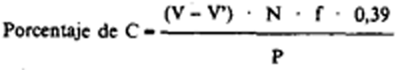
Porcentaje de extracto húmico total = porcentaje de C × 1,724
V = volumen, en ml., de sal de Mohr gastados en la prueba en blanco.
V’ = volumen, en ml., de sal de Mohr gastados en el problema.
N = normalidad de la sal de Mohr.
f = factor de la sal de Mohr.
P = peso, en gramos, de la muestra en la alícuota.
0,39 = factor que resulta de considerar que por este método sólo se oxida el 77 por 100 del carbono existente en la muestra (3).
4.4.3 Precipitación de ácidos húmicos:
Tomar 200 ml. de la solución extracto húmico total y añadir ácido sulfúrico 1:1 (4.3.6) agitando lentamente y sin turbulencias hasta pH = 1. Dejar reposar durante unas ocho horas.
Transcurrido este tiempo, centrifugar a 4.500 r.p.m. durante veinticinco minutos como máximo, para separar el precipitado de ácidos húmicos. Lavar el precipitado una vez lavado con sosa 0,5N (4.3.7), llevándolo a un matraz aforado de 50 ml. y enrasando con agua destilada.
A esta solución se la denomina «solución de ácidos húmicos».
4.4.4 Valoración de ácidos húmicos:
Tomar una parte alícuota de esta solución de ácidos húmicos, evaporar a sequedad (25 ml.) en un vaso o matraz de 500 ml., valorar siguiendo la marcha expresada en 4.4.2 y expresar el resultado como porcentaje de ácidos húmicos.
Todos los resultados se expresarán en tanto por 100 sobre el producto seco inicial.
4.6.1 Los valores orientativos para la pesada son:
|
Tipo de muestra |
Materia orgánica (materia seca) – Porcentaje |
G. de pesada (materia seca) – Gramos |
|---|---|---|
|
Abono orgánico |
35 |
07-1,5 |
|
Fertilizante órgano-mineral |
15 |
1,2-1,5 |
|
Compost |
25 |
0,7-1,2 |
|
Turba |
60 |
0,5-0,7 |
|
Enmiendas orgánicas |
15 |
1,2-1,5 |
4.6.2 Si una vez realizada la quinta repetición sigue apareciendo color oscuro, repetir modificando la pesada.
4.6.3 En el caso de utilizar bureta automática, no es necesario el empleo del ácido fosfórico ni difenilamina.
1. Determinaciones analíticas de suelos, normalización de métodos. Anales de Edafología y Agrabiología. XXXII, 1153 (1973).
2. ANNE. Ann. Agron., 15, 161-172 (1945).
3. WALKLEY, A.; BLACK, A. I., 81934). Soil Science, 37, 29.
4. KONONOVA Soil Organic Matter, Pergamon Press, 1966.
7. GRADO DE FINURA
Deshacer los agregados por simple compresión mecánica, tamizar y calcular el porcentaje que corresponda a cada tamaño, con el fin de comprobar si se ajusta a las disposiciones vigentes en la materia.
Este método es aplicable para todas aquellas materias agrupadas como productos orgánicos fertilizantes.
7.2.1 Juego de tamices circulares con un diámetro de 20 cm y de altura 5 cm.
7.2.2 Rodillo de caucho duro, taco de madera o material similar.
7.2.3 Tamizador mecánico.
Pesar, con la precisión de 0,01 g, 500 g de la muestra secada al aire y mezclar, empleando el método del cuarteo o un divisor mecánico, guardándola en un recipiente seco y hermético.
El método operatorio puede ser manual o mecánico si se dispone de un tamizador mecánico.
Tanto si se efectúa la operación a mano como mecánicamente, deben situarse los tamices de mayor a menor luz de malla.
Tomar una parte representativa de la muestra –50 a 100 g– obtenida previamente empleando el método del cuarteo y colocarla en la parte superior de la columna de tamices, es decir, en el tamiz superior.
Si se emplea el tamizador mecánico, el tiempo aproximado para efectuar la operación son diez minutos, debiéndose comprobar que hayan pasado todas las partículas en los sucesivos tamices: De no tener suficiente garantía, repetir la operación a intervalos de un minuto, hasta que hubieran pasado todas las partículas.
La operación citada también puede realizarse a mano, efectuando un movimiento circular en la columna de tamices para conseguir que las partículas vayan cruzando los sucesivos tamices.
El tamizado no debe ser prolongado más tiempo que el necesario, tratando de evitar que las partículas se disgreguen por frotación sobre los tamices.

P = peso de la muestra inicial.
Pi = peso de la muestra retenida en el tamiz de luz = i.
Expresión de los resultados:
Partículas comprendidas entre:
Luz de malla > 25: M %.
Luz de malla entre 25 y 10 mm: N %.
Luz de malla entre 10 y 5 mm: U %.
Luz de malla entre 5 y 4 mm: X %.
Luz de malla entre 4 y 1 mm: Y %.
Luz de malla < 1 mm: Z %.
Pérdidas por tamizado o error de cierre = 100 –suma distintos porcentajes (M + N + U + X + Y + Z)–.
1. Métodos Oficiales de Análisis de Fertilizantes. Ministerio de Agricultura. Madrid, 1974.
2. Ministére d l’Agriculture Belge, Méthodes de convention poru l’analyse des engrais et des emedements du sol, partie II, Amadendements du sol (1971).
8. NITRÓGENO TOTAL
Transformar el nitrógeno orgánico en sulfato de amonio por ebullición con ácido sulfúrico concentrado, previa reducción del nitrógeno nítrico a amoniacal y destilar en medio alcalino todo el N amoniacal así formado, que se valora con un ácido de titulación conocida.
8.2.1 Matraces Kjeldahl de 500 a 800 ml.
8.2.2 Aparato de destilación.
8.3.1 Ácido suflfúrico concentrado.
8.3.2 Ácido salicílico-ácido sulfúrico: Disolver 25 g de ácido salicílico en un litro de ácido sulfúrico concentrado.
8.3.3 Tiosulfato de sodio sólido.
8.3.4 Mezcla catalítica: Mezclar íntimamente 80 g de sulfato potásico, 20 g de sulfato de cobre y 2 g de selenio.
8.3.5 Solución de hidróxido de sodio al 30 por 100.
8.3.6 Solución de fenolftaleina al 1 por 100 en etanol.
8.3.7 Solución acuosa de ácido bórico al 2 por 100.
8.3.8 Indicador. Disolver 0,125 g de rojo de metilo y 0,080 g de azul de metileno en 100 ml. de etanol.
8.3.9 Ácido sulfúrico o clorhídrico 0,1 N.
Pesar de 0,2 a 2 g de la muestra (que contenga una cantidad igual o menor de 60 mg de N-nítrico), ponerlos en matraz Kjeldahl y añadir 10 ml de reactivo salicílico-sulfúrico, agitar para que se moje toda la muestra y dejar en reposo treinta minutos; añadir 1 g de tiosulfato sódico sólido y agitar; esperar quince minutos y añadir entre 10 y 15 ml de ácido sulfúrico concentrado y alrededor de 5 g de mezcla catalítica.
Colocar el matraz en el dispositivo de calentamiento que cumpla 6 (a).6.1 en cinco minutos. Calentar durante cinco minutos hasta que desaparezcan los humos blancos. Agitar suavemente por rotación y proseguir la digestión hasta que la solución se vuelva clara (lo que suele ocurrir en sesenta minutos).
Enfriar, añadir con precaución 200 ml de agua; volver a enfriar, añadir tres-cinco gotas de fenolftaleina y solución de NaOH al 30 por 100 hasta color rojo.
Inmediatamente conectar el matraz al aparato de destilación teniendo sumergido el extremo de la alargadera en un matraz Erlenmeyer o vaso conteniendo 20 ml de ácido bórico y tres-cuatro gotas de indicador.
Destilar todo el amoníaco (lo que suele ocurrir con 150 ml de destilado). Valorar el contenido del Erlenmeyer con ácido 0.1 N. El viraje es de verde a rojo burdeos.
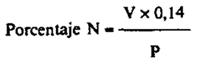
V = Volumen, en ml, de ácido 0,1 N consumidos.
P = Peso, en gramos, de la muestra.
Pueden utilizarse equipos de digestión con termorregulación y de destilación con arrastre por aire o por vapor de agua con adición semiautomática de reactivos.
Métodos oficiales de análisis de fertilizantes del Ministerio de Agricultura. Año 1977.
9. NITRÓGENO AMÍDICO (UREICO)
Este método se basa en la hidrólisis enzimática de la urea a carbonato de amonio y su valoración, previa eliminación de calcio y fosfatos.
9.2.1 Papel Albet número 242 o similar.
9.2.2 ph-metro.
9.3.1 Solución de ácido clorhídrico 0,1 N.
9.3.2 Solución de hidróxido de sodio 0,1 N.
9.3.3 Solución de ácido clorhídrico 2 N.
9.3.4 Solución de carbonato de sodio al 10 por 100.
9.3.5 Solución saturada de hidróxido de bario.
9.3.6 Ureasa liofilizada. MERCK Ref. 8.489 o equivalente. Comprobar su actividad enzimática periódicamente y guardar a temperatura inferior a 4 ° C. Utilizarla en suspensión recién preparada.
9.3.7 Disolución neutra de ureasa. Preparar una suspensión de ureasa en agua destilada al 0,25 por 100 y neutralizar a pH = 4,4.
Tomar 10 g de muestra, pesados con precisión de 1 mg e introducirlos en un matraz aforado de 250 ml, llevar a volumen con agua destilada, agitar quince minutos y filtrar sobre papel Albet 242 o similar. Tomar con pipeta 50 ml de la solución filtrada y pasarlos a un matraz aforado de 500 ml
Añadir suficiente solución saturada de hidróxido de bario para precipitar los fosfatos, dejar sedimentar y comprobar si la precipitación fue completa.
Añadir solución de carbonato de sodio para precipitar el exceso de bario y cualquier sal de calcio soluble. Dejar sedimentar y comprobar de nuevo si la precipitación fue completa.
Mezclar y llevar a volumen. Filtrar sobre papel seco Albet 242 o similar y trasvasar 50 ml de filtrado a un matraz Erlenmeyer de 250 ml, neutralizar con solución de ácido clorhídrico 2 N y añadir dos o tres gotas en exceso. Neutralizar la disolución con hidróxido de sodio 0,1 N hasta pH = 4,4.
Añadir a cada muestra 20 ml de suspensión de eureasa, tapar con tapón de goma y dejar en reposo durante una hora a 20-25 ° C. Enfriar a 0 ° C y valorar con ácido clorhídrico 0,1 N añadiendo un exceso de este reactivo y valorar por retroceso con hidróxido de sodio 0,1 N hasta pH = 4,4. Anotar el volumen total, en ml añadido de ácido clorhídrico 0,1 N(A) y de hidróxido de sodio 0,1 N(B).
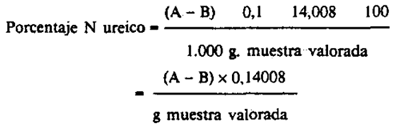
Métodos de análisis AOAC, 13.ª edición, 1980, números 2.080 y 2.081.
10. NITRÓGENO NÍTRICO
(Método de Roberstson)
Determinar el N total y el N insoluble en agua. La diferencia entre ambos es el N soluble.
En la disolución de Nd soluble eliminar el N nítrico al estado de óxido nítrico por medio de sulfato de hierro (II). Una vez eliminado, determinar el N total en el residuo y la diferencia entre el N soluble y este último es el N nítrico.
Aplicable en presencia de cianamida cálcica y urea.
10.2.1 Matraces Kjeldahl de 500 a 800 ml.
10.2.2 Aparatos de destilación.
10.3.1 Sulfato de hierro (II) heptahidrato (Fe SO47H2O).
10.3.2 Óxido de mercurio o mercurio metálico, exento de N.
10.3.3 Sulfato de potasio o de sodio anhidro, exento de N.
10.3.4 Ácido sulfúrico de 93 a 98 por 100, exento de N.
10.3.5 Disolución de tiosulfato de sodio o de sulfuro de sodio: 80 g de S2O3Na2 · 5H2O en 1 litro de agua o 40 g de sulfuro de sodio en 1 litro de agua.
10.3.6 Hidróxido de sodio o en disolución: 450 g de NaOH en agua, enfriar y enrasar a 1 litro. Debe tener de densidad 1,36 o más.
10.3.7 Regulador de ebullición inerte.
10.3.8 Rojo de metilo: Disolver 1 g en 100 ml de etanol.
10.3.9 Disolución de ácido sulfúrico o clorhídrico 0,1 N.
10.3.10 Disolución de hidróxido de sodio 0,1 N.
10.4.1 Modalidad A: Caso general en que es necesario determinar el N insoluble en agua.
Determinar el N total por el método oficial 8.
10.4.2 Separar y determinar el N insoluble en agua por el método correspondiente.
10.4.3 En la disolución obtenida en 10.4.2, eliminar el N nítrico y determinar el N restante.
Para ello, colocar el filtrado procedente del apartado anterior en un matraz Kjeldahl de 500 ml y añadir 2 g de SO2Fe . 7H2O y 20 ml.
10.4.4 Ponerlo a la llama hasta evaporar el agua y que aparezcan humos blancos. Continuar la digestión por lo menos diez minutos más para expulsar todo el N nítrico. Si se produce una fuerte vaporización, añadir 10 ó 15 perlas de vidrio.
10.4.5 Agregar 0,65 g de Hg ó 0,7 de HgO y continuar la digestión hasta que toda la materia orgánica se haya oxidado.
Determinar el N total por el método oficial 8.
10.4.6 Modalidad B: Modificación de Jones para el caso en que no hace falta determinar el N insoluble en agua por ser todo él soluble.
Determinar el N total por el método oficial 8.
10.4.7 Eliminar el N nítrico y determinar el N restante.
Para ello pesar 0,5 g del problema, colocarlos en un matraz Kjeldahl de 500 ml, añadir 50 ml de agua y agitar suavemente.
Agregar 2 g de SO4Fe . 7H2O y 20 ml de SO4H2, continuando como en 10.4.4 y 10.4.5.
Modalidad A:
N total – N insoluble = N soluble.
N soluble – N obtenido en 10.4.5 = N nítrico.
Modalidad B:
N total antes de eliminar el N nítrico menos N total después de su eliminación = N nítrico.
Método A.O.A.C. edición 1970, números 2.061 y 2.062.
12. NITRÓGENO ORGÁNICO
Este método es muy semejante al de Robertson, salvo que al final se determinan también el N amoniacal y el N ureico para deducirlos, junto con el N nítrico, del N total.
Matraces Kjeldahl de 500 a 800 ml.
Como en 8.3, 9.3 y 10.3 métodos de N total, N amídico y N nítrico.
12.4.1 Determinar el N total por el método oficial número 8.
12.4.2 Separar y determinar el N insoluble en agua por el método correspondiente.
12.4.3 En sendas porciones del filtrado procedente del apartado anterior, determinar el N nítrico (por el método de Robertson), el N amoniacal y el N ureico (por el método de la ureasa).
N orgánico = T – (N + A + U)
T = N total.
N = N nítrico.
A = N amoniacal.
U = N ureico.
Métodos de la A.O.A.C. edición 1970, números 2.051 y siguiente.
14. FÓSFORO SOLUBLE EN AGUA Y EN CITRATO AMÓNICO
(Asimilable)
Después de separar el fósforo solubre en agua de la muestra, someter el residuo a una extracción con disolución neutra (pH = 7,0) de citrato amónico. El fósforo asimilable puede determinarse en la disolución obtenida al reunir los extractos acuosos y de citrato amónico.
Aplicable a aquellos abonos en que se exige el fósforo solubre en agua y en citrato conjuntamente, tales como los abonos organominerales.
14.2.1 Un pH-metro.
14.2.2 El resto como en 13.2.
14.2.3 Crisol filtrante de 5-15 u, G4.
14.3.1 Citrato amónico neutro.
Debe tener un peso específico de 1,09 a 20° C y un pH igual 7,0 determinado electrométricamente.
Disolver 370 g de ácido cítrico cristalizado en 1,5 litros de agua y casi neutralizar añadiendo 345 ml de hidróxido de amonio (de 28 a 29 por 100 de NH3). Si la concentración de amoniaco es menos del 28 por 100, añadir mayor volumen y disolver el ácido cítrico en un volumen más pequeño de agua. Enfriar y comprobar el pH. Ajustar con hidróxido de amonio (1:7) o con disolución de ácido cítrico a pH = 7,0. Si es preciso, diluir la disolución para que el peso específico sea 1,09 a 20 C. El volumen será aproximadamente de 2 litros. Conservar en frascos herméticamente cerrados y comprobar el pH de cuando en cuando, reajustándolo si difiere de 7,0.
14.3.2 Reactivo quimociaco. Como en 13.3.4.
14.4.1 Fósforo soluble en agua: Colocar 1 g de muestra en un filtro de 9 cm de diámetro y lavar con pequeñas porciones de agua hasta que el filtrado mida algo menos de 250 ml.
Dejar pasar cada porción a través del filtro antes de añadir más. Si el lavado no puede completarse al cabo de una hora, utilizar succión. En caso de que el filtrado esté turbio, añadir 1 ó 2 ml de NO3H concentrado. Enrasar a 250 ml con agua y agitar (disolución I).
14.4.2 Preparación de las disoluciones: Después de retirar el P2O5 soluble en agua (disolución I), transferir el filtro y residuo, en un tiempo no superior a una hora, a un matraz de 250 ml que contenga 100 ml de la disolución de citrato amónico neutro.
Cerrar el matraz herméticamente con tapón de goma compacto, sacudir enérgicamente hasta que el papel se reduzca a pulpa y reducir la presión quitando momentáneamente el tapón.
Dejar en reposo el matraz durante veinticuatro horas.
14.4.3 Añadir el filtro y el residuo y filtrar el contenido por succión, tan rápidamente como sea posible, a través del papel Whatman número 5 o equivalente utilizando un buchner.
Lavar con agua hasta el que el volumen del filtrado sea aproximadamente de 200 ml, dejando tiempo para que el drenaje sea completo antes de añadir más agua. Si el material es tal que produce filtrado turbio, lavar con disolución de nitrato de amonio al 5 por 100. Enfriar y enrasar (disolución II).
Tomar una alícuota de la disolución I y otra de la disolución II, ponerlas en un erlenmeyer de 400 ml (entre las dos alícuotas no deben tener más de 25 mg de P205) y diluir a 50 ml si es necesario.
14.4.4 Método gravimétrico como fosfomolibdato de quinoleína: Añadir 50 ml del reactivo quimociaco, cubrir con vidrio de reloj, colocar en placa caliente con vitrina bien ventilada y hervir durante un minuto, enfriar a temperatura ambiente, agitar cuidadosamente formando remolinos tres o cuatro veces durante el enfriamiento.
Filtrar con un crisol (13.2.1) previamente desecado a 250° C y tarado, lavar cinco veces con porciones de 25 ml de agua.
Desecar el crisol con su contenido a 250° C durante treinta minutos, enfriar en desecador y pesar el fosfomolibdato de quinoleína (C9H7N3) H3 (PO4 12MoO3). Efectuar paralelamente un ensayo en blanco.
Como en 13.5.
Como en 13.6.
14.7.1 Métodos oficiales A.O.A.C. edición 1970, números 2.032, 2.025, 2.037 y 2.038.
15. NITRÓGENO INSOLUBLE EN AGUA
Separar en un filtro la parte de abono que no se solubiliza en agua y determinar en ella el N total.
Aplicar cuando se pide separadamente el N soluble y el insoluble, como en los abonos y enmiendas orgánicos.
15.2.1 Como en el 8.2.
15.2.2 Filtro Whatman número 2 o similar, de 12 cm de diámetro.
15.3.1 Etanol del 95 por 100.
15.3.2 El resto como en 8.3.
15.4.1 Pesar 2,5 g de la muestra y colocar en un erlenmeyer de 50 ml, humedeciéndola con etanol. Añadir 20 ml de agua y esperar quince minutos agitando de cuando en cuando.
Filtrar al vacío decantando por el filtro (15.2.2).
Lavar cuatro o cinco veces con agua a la temperatura ambiente (20-25° C) y decantar.
Finalmente pasar todo el residuo al filtro.
15.4.2 Recoger el filtro con el residuo, introducirlo en un matraz Kjeldahl y determinar en ello el N total como en el método Kjeldahl para muestras sin nitratos.
Como en 8.5.
Método A.O.A.C. edición 1980, número 2.064.
17. POTASIO TOTAL
Consiste en solubilizar el potasio insoluble en agua, pero que puede ser útil como fertilizante, y determinar el total a continuación según el método número 16.
17.2.1 Cápsulas de platino o porcelana.
17.2.2 Como en 16.
17.3.1 Disolución de ácido clorhídrico al 1 por 100.
17.3.2 Ácido nítrico concentrado.
17.3.3 El resto, como en 16.3.
Pesar 10 g del problema y calcinar aproximadamente a 500° C en una cápsula (17.2.1) lentamente, removiendo el contenido y no pasando de esa temperatura. Obtenida la calcinación, agregar a la cápsula agua caliente, decantando cuidadosamente el contenido a un erlenmeyer de 250 ml.
Volver a agregar a la cápsula agua acidulada con CLH (17.3.1), calentando con cuidado para que no haya proyecciones, lavar con agua caliente y agregar cinco gotas de NO3H arrastrando todo el contenido de la cápsula al erlenmeyer en el que debe hervir el contenido durante unos diez minutos. Dejar enfriar a la temperatura ambiente, pasar el contenido a un matraz aforado de 250 ml, enrasar y homogeneizar.
Filtrar, y del filtrado tomar 50 ml, que equivalen a 2 g de problema, y evaporar en la cápsula hasta sequedad, calcinando ligeramente.
El residuo se trata por agua caliente y filtra, lavando escrupulosamente la cápsula y el filtro.
En el filtrado, determinar el potasio según el método número 16.
Como en 16.5.
Como en 16.6.
Métodos Oficiales de Análisis de Fertilizantes. Ministerio de Agricultura. Año 1981.
18. AMINOÁCIDOS LIBRES Y TOTALES
El método está basado en la separación y determinación de los distintos aminoácidos por cromatografía líquida de alta presión.
Previamente a la cromatografía los aminoácidos reaccionan con el reactivo OPA (Ortoftalaldehído) y FNIC - Cl (fluorenil metil cloro formiato) para formar derivados fluorescentes (Derivación Precolumna).
18.2.1 Tubos de vidrio de 30 ml, tipo SOVIREL, o similar con tapón a rosca.
18.2.2 Matraces de fondo redondo, de aproximadamente 100 ml, de boca esmerilada.
18.2.3 Desecador con dos bocas.
18.2.4 Bomba de vacío.
18.2.5 Estufa de desecación con regulación automática de temperatura.
18.2.6 Evaporador rotatorio.
18.2.7 Papel de filtro Albet número 240, 242 o similar.
18.2.8 Matraces aforados de 50 ml.
18.2.9 Tubos de 10 ml.
18.2.10 Viales de 10 ml.
18.2.11 Cromatógrafo líquido de alta presión con detector de fluorescencia.
Condiciones de detección con OPA: Longitud de onda de excitación 340 nm. Longitud de onda de detección: 420 nm.
Condiciones de detección con FMOC: Longitud de onda de excitación: 250 nm. Longitud de onda de detección: 335 nm.
18.2.12 Columna C 18, de 5 u de tamaño de partícula y de 20 cm de longitud.
18.2.13 Fase móvil:
18.3.9 Reactivo FMOC:
Disolver 155 mg de FMOC en 40 ml de acetona.
18.3.10 Hidróxido sódico 0,1 N.
18.3.11 Nitrógeno (gas puro).
18.3.12 Patrón de aminoácidos para calibración:
Preparar una disolución patrón de aminoácidos conteniendo las siguientes cantidades de cada uno de ellos, en ácido clorhídrico 0,1 N.
|
|
ug/ml |
|---|---|
|
Ácido aspártico |
6 |
|
Ácido glutámico |
– |
|
Serina |
– |
|
Histedina |
8 |
|
Glicina |
4 |
|
Treonina |
6 |
|
Arginina |
4 |
|
Alanina |
4 |
|
Tirosina |
8 |
|
Metionina |
8 |
|
Valina |
6 |
|
Fenilalanina |
8 |
|
Isoleucina |
6 |
|
Prolina |
6 |
|
Leucina |
6 |
|
Lisina |
6 |
18.4.1 Aminoácidos totales: Hidrólisis con ácido clorhídrico 6 N. Pesar una cantidad de muestra (que contengan aproximadamente 1,5 mg de nitrógeno procedente de aminoácidos) e introducirla en un tubo de vidrio de 30 ml provisto de tapón de rosca (18.2.1).
Agregar 15 ml de la disolución de fenol en ácido clorhídrico 6 N (18.3.1).
Llevar a un dispositivo donde pueda inyectarse nitrógeno para disponer de una atmósfera exenta de oxígeno (desecador con dos bocas, por una de las cuales se inyecta nitrógeno y por la otra se extrae con una bomba de vacío).
Esta operación se realiza cinco veces.
Llevar a una estufa de 100-105 ° C durante veinticuatro horas.
Evaporar en rotovapor a una temperatura de 40-50 ° C.
Una vez obtenido el residuo se diluye con 25 ml de agua destilada y se vuelve a evaporar (en caso de seguir oliendo a ácido clorhídrico se repite la operación).
Arrastrar el residuo con agua destilada, filtrar sobre papel de filtro seco y recoger el filtrado en un matraz de 50 ml y aforar.
18.4.2 Aminoácidos libres.
Pesar la cantidad de muestra necesaria (que contenga aproximadamente 1,5 mg de nitrógeno procedente de aminoácidos) en un matraz de 50 ml y diluir hasta volumen con ácido clorhídrico 0,1 N.
18.4.3 Derivación.
Mezclar en un tubo 1 ml de solución de la muestra obtenida según (18.4.1 y 18.4.2) con 0,1 ml de hidróxido sódico 0,1 N y 2,9 ml de agua destilada.
Pasar 100 ul de esta disolución a un vial, añadir 100 ul de reactivo OPA y mezclar. Esperar un minuto. Añadir 100 ul del reactivo FMOC. Esperar cuarenta segundos. Añadir 100 ul de pentano y agitar. Dejar separar las fases e inyectar 10 ul de la fase acuosa en el cromatógrafo.
18.4.4 Cromatografía.
Las longitudes de onda de iniciación del detector son:
Exc. 340 nm.
Condiciones de detección con OPA.
Em. 420 nm.
Estas condiciones se mantienen hasta que se obtenga el pico correspondiente a la isoleucina e inmediatamente se cambian las longitudes de onda a:
Exc. 250 nm.
Condiciones de detección con FMOC.
Em. 335 nm.
Después de la elución del pico correspondiente a la prolina se vuelve a cambiar de nuevo las longitudes de onda a:
Exc. 340 nm.
Condiciones de detección con OPA.
Em. 420 nm.
Los resultados se expresarán en porcentaje (P/P) por comparación de las áreas del pico de cada aminoácido con el correspondiente en el cromatograma de la solución patrón.
En caso de efectuarse hidrólisis los resultados del triptofano no son exactos y la sensibilidad para la cistina es baja.
El triptofano se determinaría sometiendo la muestra a una hidrólisis alcalina.
La cistina sometiendo la muestra a una oxidación con ácido perfórmico previa a la hidrólisis.
18.7.1 Davies, M.G.; Thomas, A.J. «An Investigation of Hidrolytic Techniques for the Aminoacid Analysis of Foods tuffs». J. Sci. Fd. Agric. 1973, 24, 1541-1550.
18.7.2 Pfeifer, R.; Korpi, J.; Burgoyne, R.; Mc Court, D. «Practical Application of HPLC to aminoacid analysis». American Laboratory March, 1983.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




